

Deep sequencing of SMPD1 gene revealed a heterozygous frameshift mutation (p.Ser192Alafs) in a Palestinian infant with Niemann-Pick disease type A: a case report
- PMID: 30223864
- PMCID: PMC6142321
- DOI: 10.1186/s13256-018-1805-x
Deep sequencing of SMPD1 gene revealed a heterozygous frameshift mutation (p.Ser192Alafs) in a Palestinian infant with Niemann-Pick disease type A: a case report
Abstract
Background: Niemann-Pick disease is caused by reduced level of the lysosomal enzyme acid sphingomyelinase. Children can survive between 2 and 12 years based on the disease type. Two main types are well known: type A and B. Niemann-Pick disease type A is characterized by severe central nervous system deterioration and hepatosplenomegaly while type B is a progressive hypersplenism accompanied with gradual deterioration of pulmonary function.
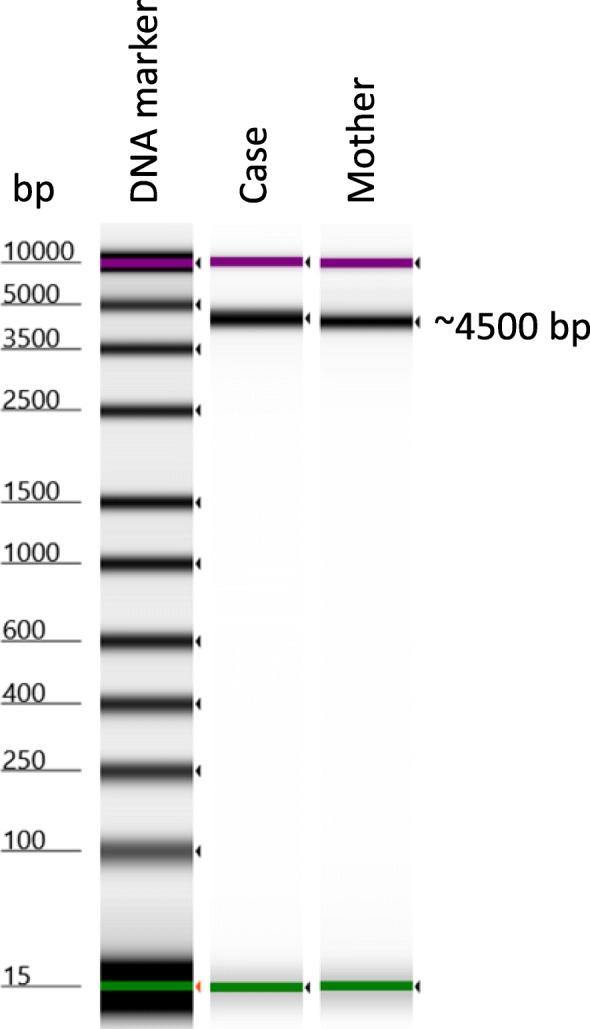
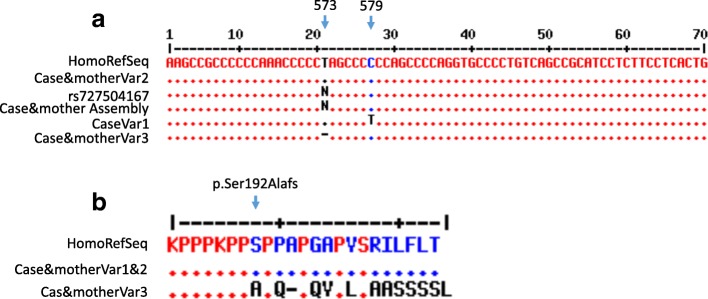
Case presentation: We describe an 11-month-old Palestinian baby boy with hepatosplenomegaly, hypotonia, delayed motor development, laryngomalacia, bilateral cherry-red spots, and failure to thrive. Metabolic screening, blood count, differential tests, immunology screen, infectious disease screen, urine, biochemical tests as well as molecular diagnosis were performed. The molecular diagnosis was done by amplifying the whole sphingomyelin phosphodiesterase 1 (SMPD1) gene, followed by deep sequencing. The obtained sequences were aligned, de novo assembled and compared to human reference gene (GenBank GeneID: NG_011780.1, Ensembl version ENSG00000166311 and protein identified as UniProtKB - P17405). Two known mutations were identified in our patient: the pathogenic frameshift mutation NM_000543.4(SMPD1):c.573delT (p.Ser192Alafs) and the benign polymorphism NM_000543.4(SMPD1):c.107T>C (p.Val36Ala). The enzyme study showed a very low level of enzymatic activity of acidic sphingomyelinase (0.1 nmol/ml per hour). Correlations between clinical findings, laboratory data, and sequence analysis are presented.
Conclusions: In conclusion, this is the first report about a heterozygote frameshift p.Ser192AlafsX65 in a Palestinian patient with Niemann-Pick disease type A, emphasizing the importance of deep sequencing in genetic diagnosis of this rare inherited disease.
Keywords: Mutation; Niemann–Pick disease type A; Palestinian child; Sphingomyelinase deficiency.
Conflict of interest statement
Ethics approval and consent to participate
The study protocol was approved by the ethics committee of Al-Quds University Ref. no. 25/REC/2017.
Consent for publication
Written informed consent was obtained from the patient’s legal guardian(s) for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Schuchman EH, Wasserstein MP. Types A and B Niemann-Pick disease. Pediatr Endocrinol Rev. 2016;13(Suppl 1):674–681. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
