

Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities
- PMID: 30227272
- PMCID: PMC6309415
- DOI: 10.1016/j.freeradbiomed.2018.09.019
Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities
Abstract
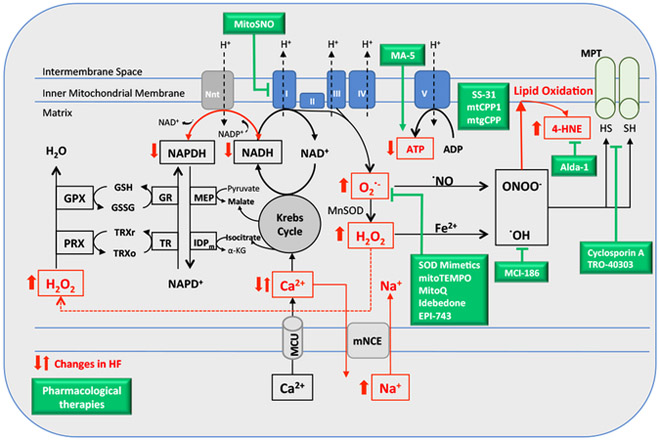
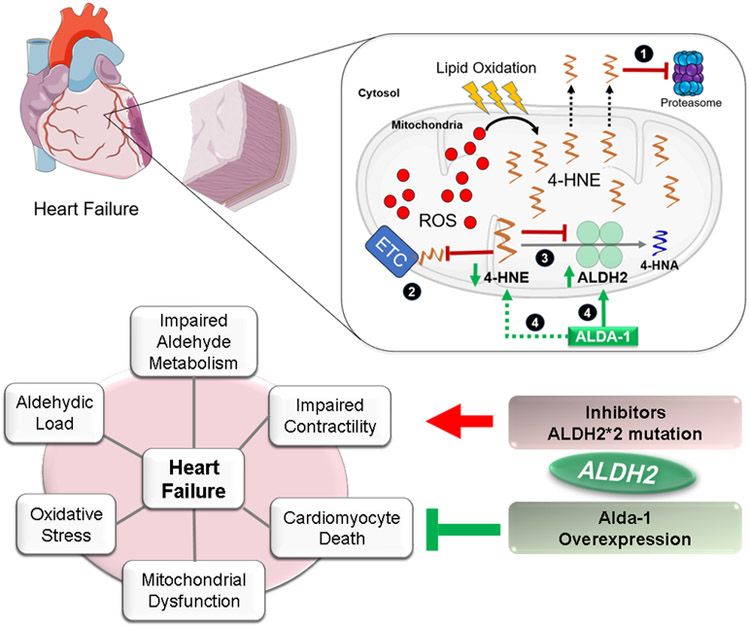
Mitochondrial dysfunction characterized by impaired bioenergetics, oxidative stress and aldehydic load is a hallmark of heart failure. Recently, different research groups have provided evidence that selective activation of mitochondrial detoxifying systems that counteract excessive accumulation of ROS, RNS and reactive aldehydes is sufficient to stop cardiac degeneration upon chronic stress, such as heart failure. Therefore, pharmacological and non-pharmacological approaches targeting mitochondria detoxification may play a critical role in the prevention or treatment of heart failure. In this review we discuss the most recent findings on the central role of mitochondrial dysfunction, oxidative stress and aldehydic load in heart failure, highlighting the most recent preclinical and clinical studies using mitochondria-targeted molecules and exercise training as effective tools against heart failure.
Keywords: Aldehydes; Cardiovascular diseases; Exercise training; Mitochondria; Redox imbalance; Therapy.
Copyright © 2018 Elsevier Inc. All rights reserved.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
