

Targeting nonsense-mediated mRNA decay in colorectal cancers with microsatellite instability
- PMID: 30228267
- PMCID: PMC6143633
- DOI: 10.1038/s41389-018-0079-x
Targeting nonsense-mediated mRNA decay in colorectal cancers with microsatellite instability
Abstract
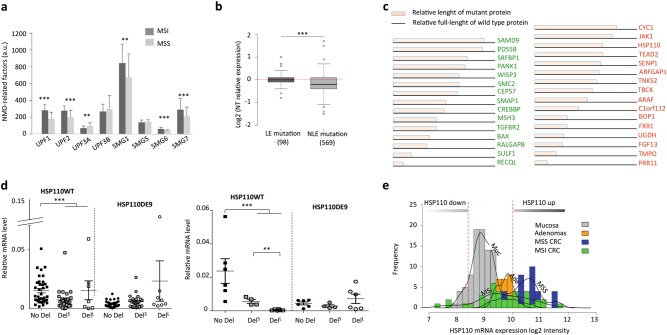
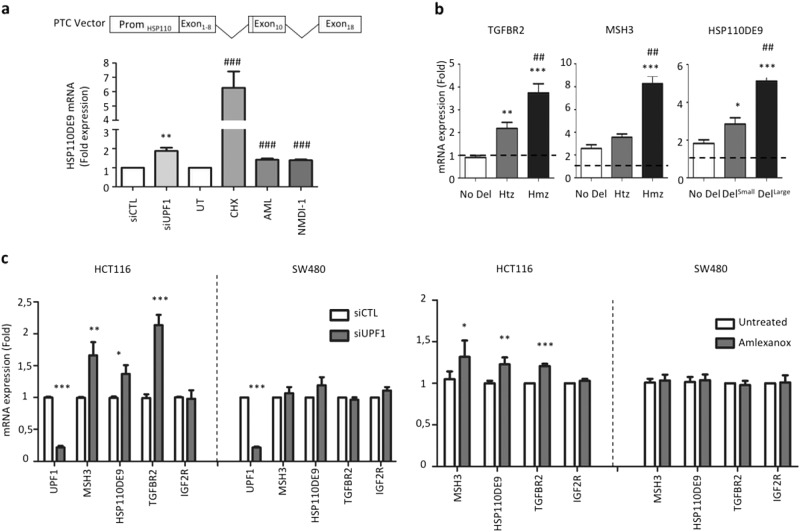
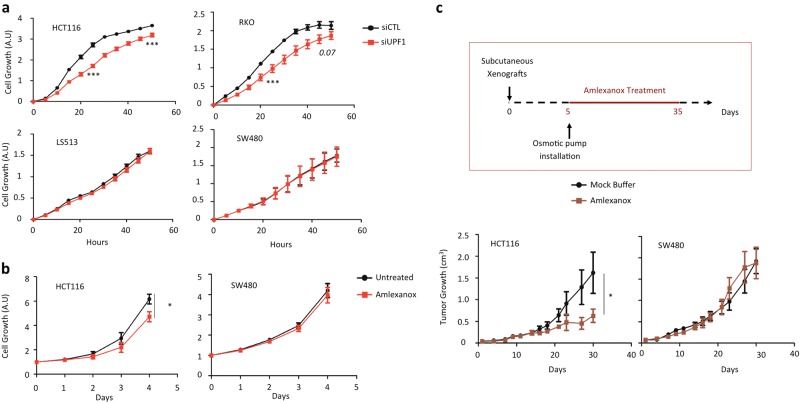
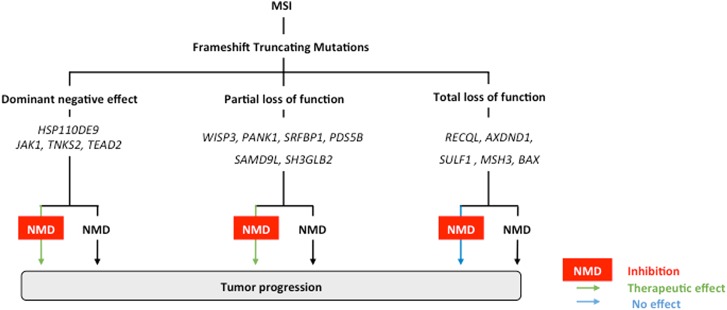
Nonsense-mediated mRNA decay (NMD) is responsible for the degradation of mRNAs with a premature termination codon (PTC). The role of this system in cancer is still quite poorly understood. In the present study, we evaluated the functional consequences of NMD activity in a subgroup of colorectal cancers (CRC) characterized by high levels of mRNAs with a PTC due to widespread instability in microsatellite sequences (MSI). In comparison to microsatellite stable (MSS) CRC, MSI CRC expressed increased levels of two critical activators of the NMD system, UPF1/2 and SMG1/6/7. Suppression of NMD activity led to the re-expression of dozens of PTC mRNAs. Amongst these, several encoded mutant proteins with putative deleterious activity against MSI tumorigenesis (e.g., HSP110DE9 chaperone mutant). Inhibition of NMD in vivo using amlexanox reduced MSI tumor growth, but not that of MSS tumors. These results suggest that inhibition of the oncogenic activity of NMD may be an effective strategy for the personalized treatment of MSI CRC.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res. 2002;62:2447–2454. - PubMed
-
- Duval A, et al. The human T-cell transcription factor-4 gene: structure, extensive characterization of alternative splicings, and mutational analysis in colorectal cancer cell lines. Cancer Res. 2000;60:3872–3879. - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
