

Evolution of gene knockout strains of E. coli reveal regulatory architectures governed by metabolism
- PMID: 30228271
- PMCID: PMC6143558
- DOI: 10.1038/s41467-018-06219-9
Evolution of gene knockout strains of E. coli reveal regulatory architectures governed by metabolism
Abstract
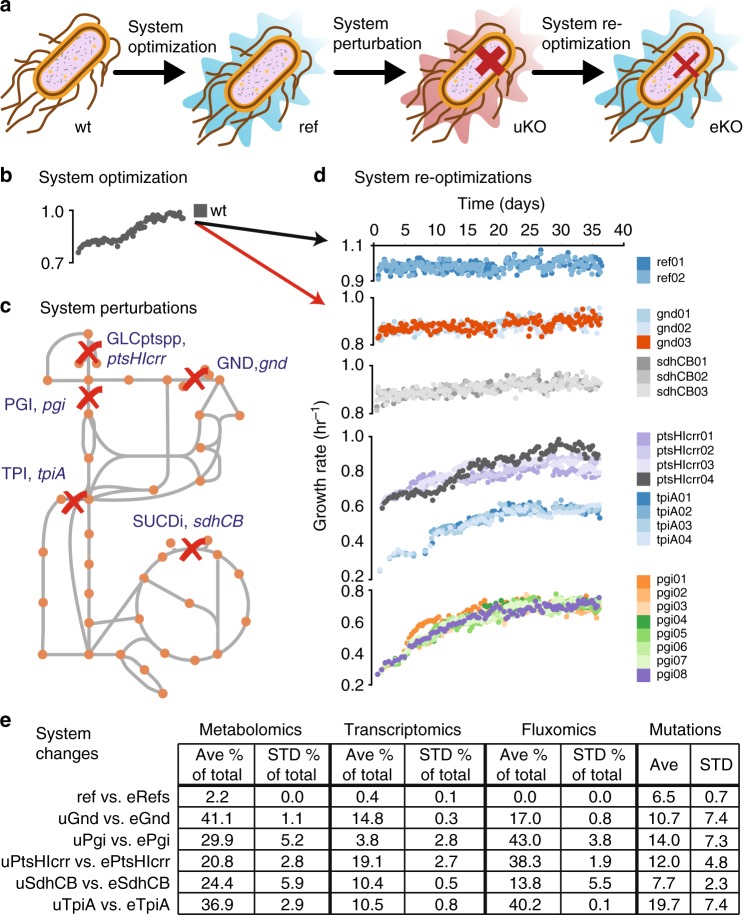
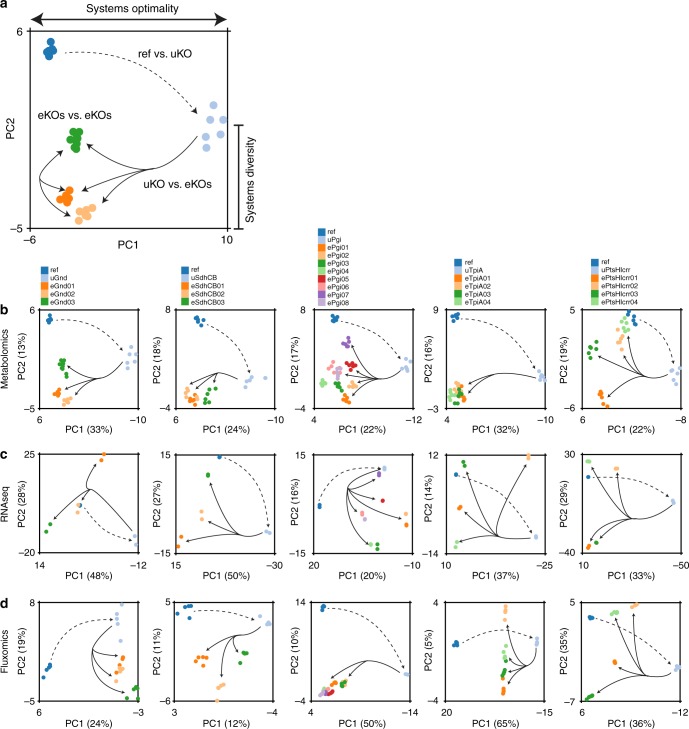
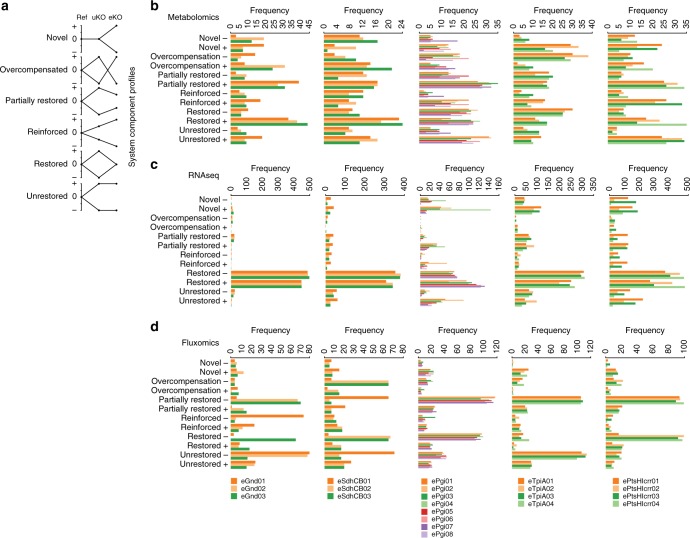
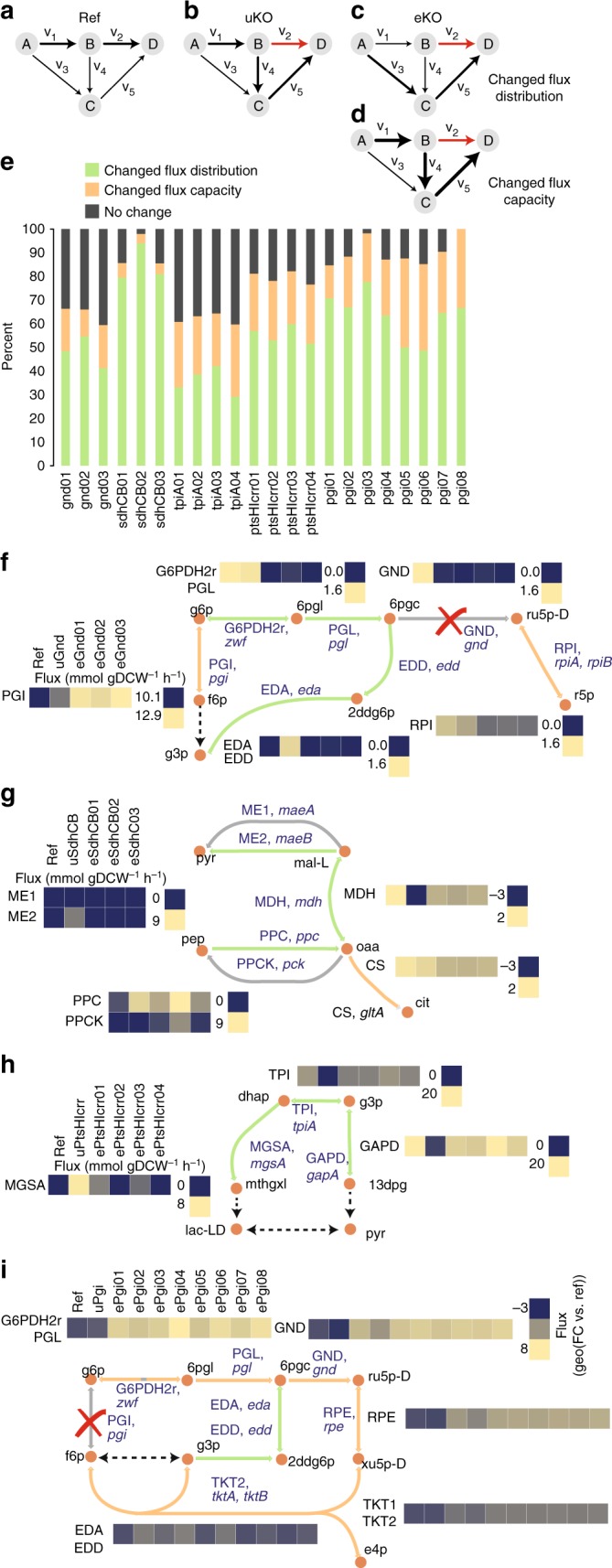
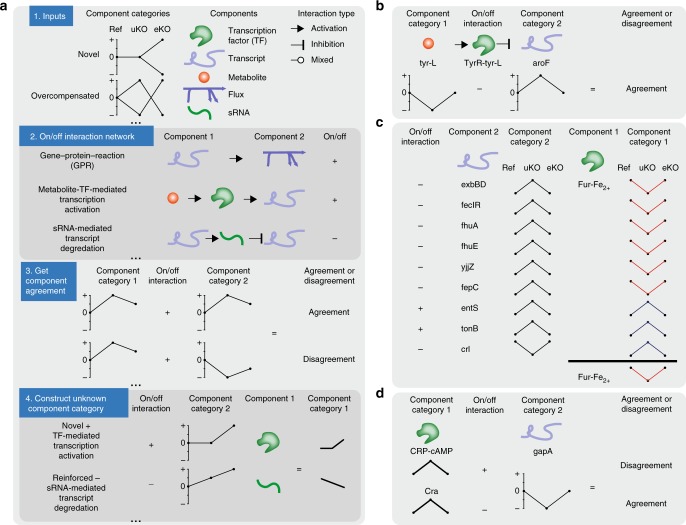
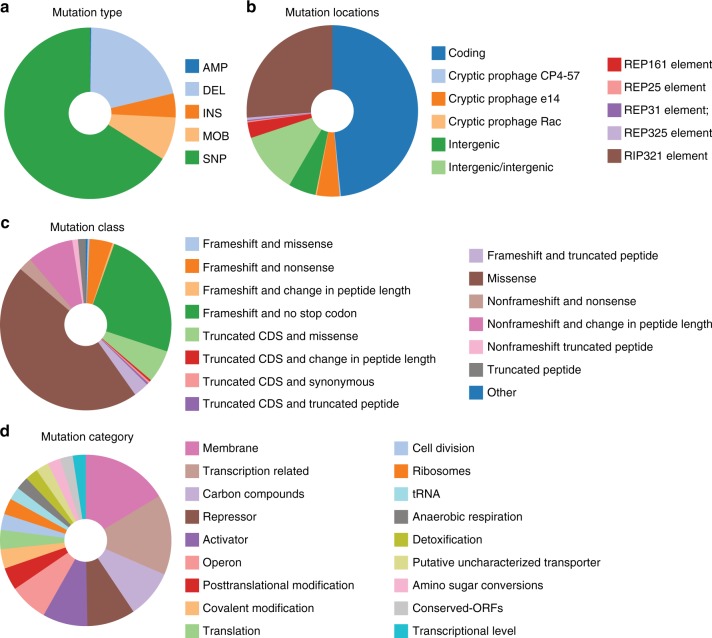
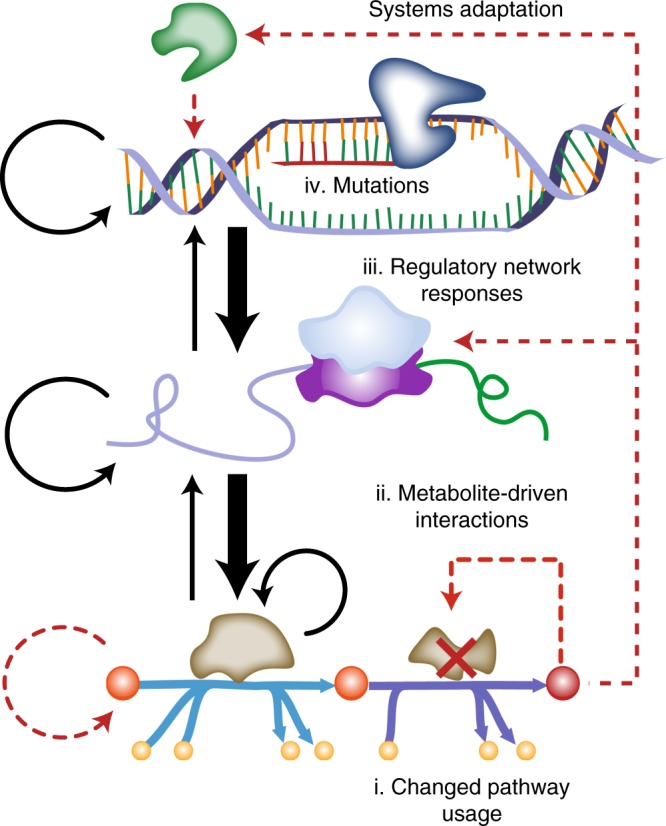
Biological regulatory network architectures are multi-scale in their function and can adaptively acquire new functions. Gene knockout (KO) experiments provide an established experimental approach not just for studying gene function, but also for unraveling regulatory networks in which a gene and its gene product are involved. Here we study the regulatory architecture of Escherichia coli K-12 MG1655 by applying adaptive laboratory evolution (ALE) to metabolic gene KO strains. Multi-omic analysis reveal a common overall schema describing the process of adaptation whereby perturbations in metabolite concentrations lead regulatory networks to produce suboptimal states, whose function is subsequently altered and re-optimized through acquisition of mutations during ALE. These results indicate that metabolite levels, through metabolite-transcription factor interactions, have a dominant role in determining the function of a multi-scale regulatory architecture that has been molded by evolution.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
