

Biosynthesis, structure, and folding of the insulin precursor protein
- PMID: 30230185
- PMCID: PMC6463291
- DOI: 10.1111/dom.13378
Biosynthesis, structure, and folding of the insulin precursor protein
Abstract
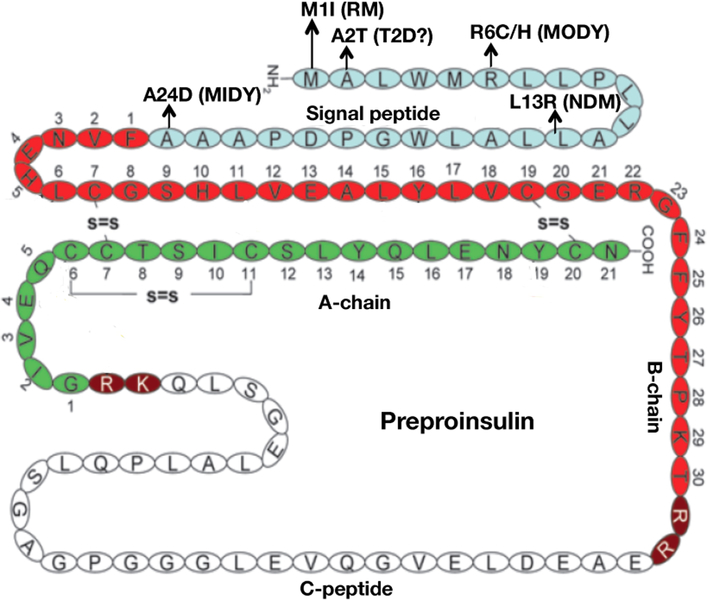
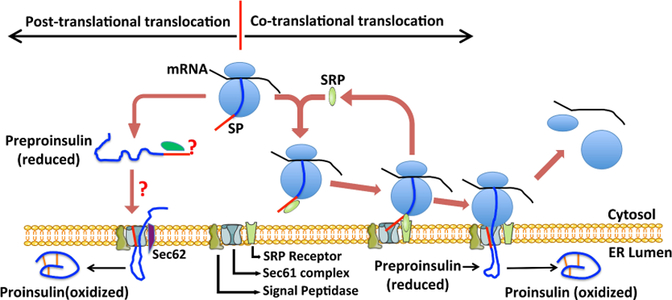
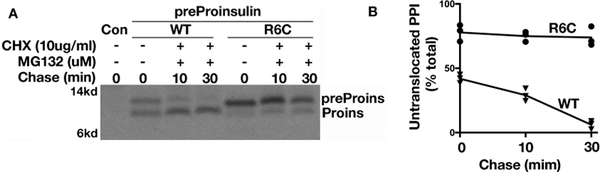
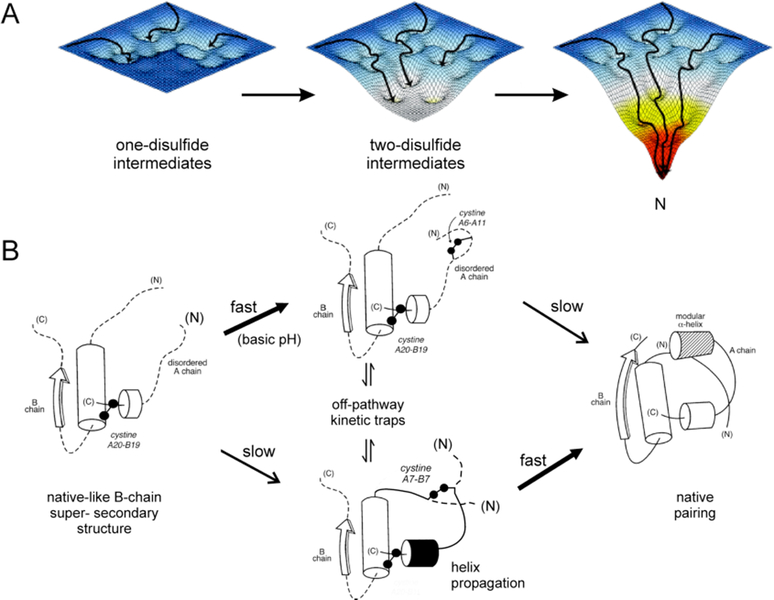
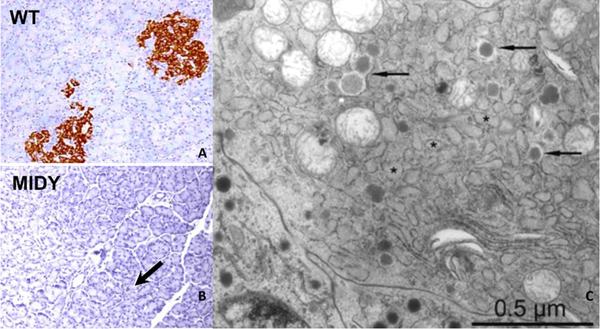
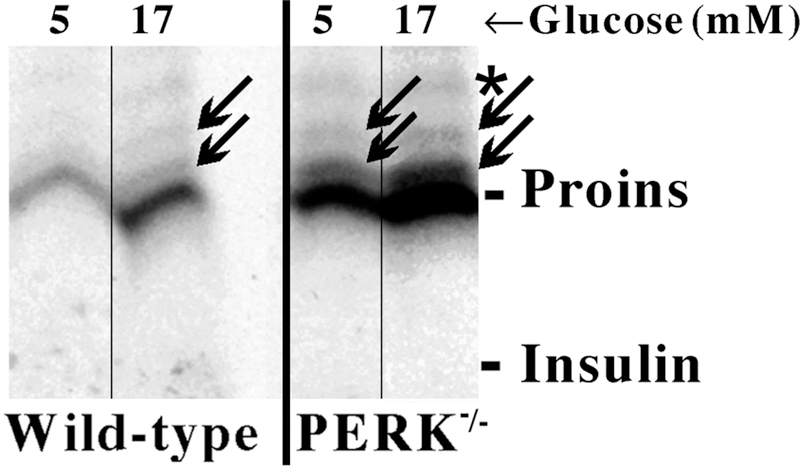
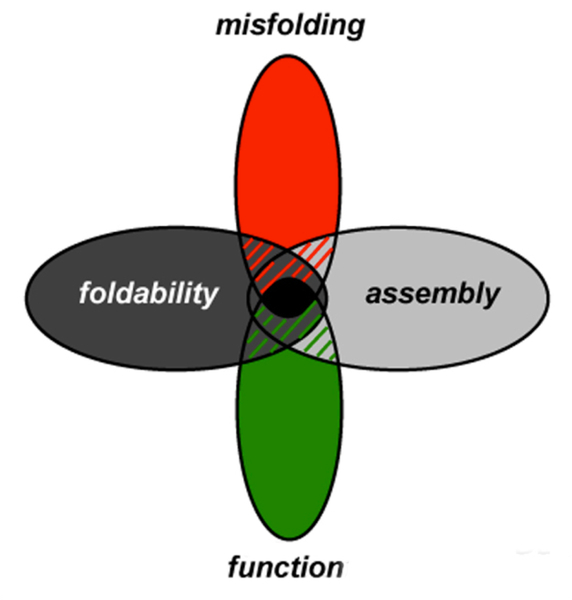
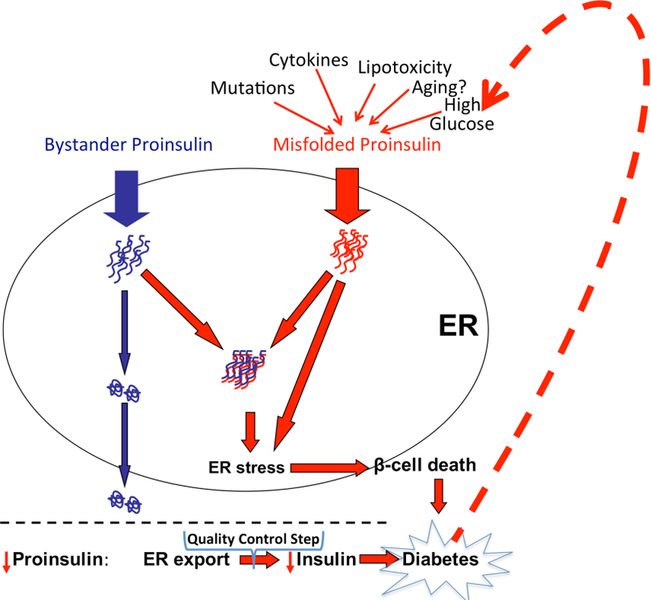
Insulin synthesis in pancreatic β-cells is initiated as preproinsulin. Prevailing glucose concentrations, which oscillate pre- and postprandially, exert major dynamic variation in preproinsulin biosynthesis. Accompanying upregulated translation of the insulin precursor includes elements of the endoplasmic reticulum (ER) translocation apparatus linked to successful orientation of the signal peptide, translocation and signal peptide cleavage of preproinsulin-all of which are necessary to initiate the pathway of proper proinsulin folding. Evolutionary pressures on the primary structure of proinsulin itself have preserved the efficiency of folding ("foldability"), and remarkably, these evolutionary pressures are distinct from those protecting the ultimate biological activity of insulin. Proinsulin foldability is manifest in the ER, in which the local environment is designed to assist in the overall load of proinsulin folding and to favour its disulphide bond formation (while limiting misfolding), all of which is closely tuned to ER stress response pathways that have complex (beneficial, as well as potentially damaging) effects on pancreatic β-cells. Proinsulin misfolding may occur as a consequence of exuberant proinsulin biosynthetic load in the ER, proinsulin coding sequence mutations, or genetic predispositions that lead to an altered ER folding environment. Proinsulin misfolding is a phenotype that is very much linked to deficient insulin production and diabetes, as is seen in a variety of contexts: rodent models bearing proinsulin-misfolding mutants, human patients with Mutant INS-gene-induced Diabetes of Youth (MIDY), animal models and human patients bearing mutations in critical ER resident proteins, and, quite possibly, in more common variety type 2 diabetes.
Keywords: Sec61 translocon; disulphide-linked protein complexes; polypeptide chain initiation; secretory protein biosynthetic pathway; unfolded protein response.
© 2018 John Wiley & Sons Ltd.
Conflict of interest statement
Conflict of interest
None declared.
Figures
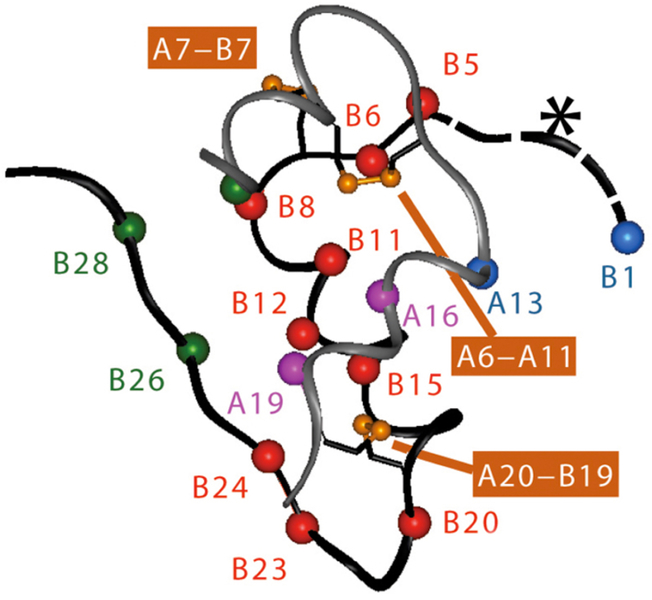
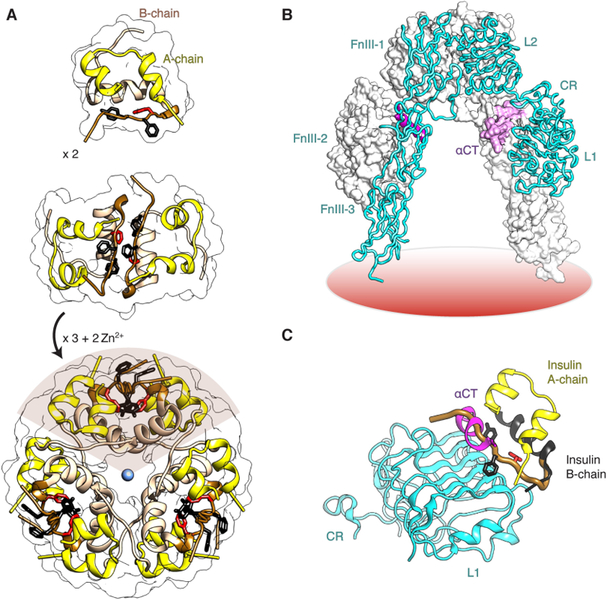
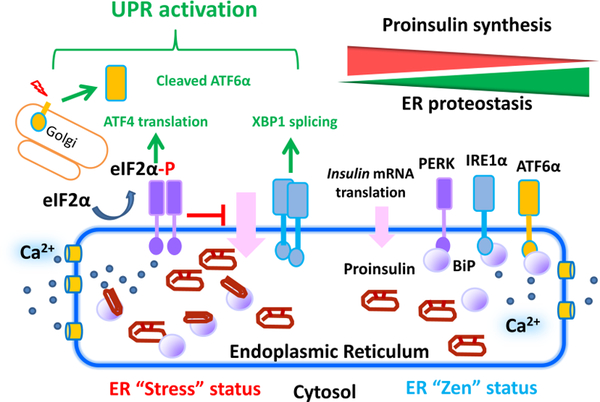
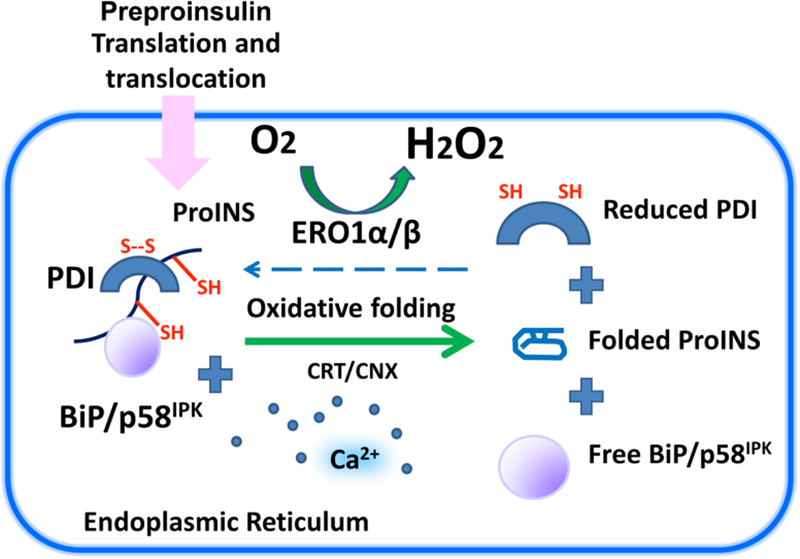
References
-
- Gold G, Gishizky ML, Grodsky GM. Evidence that glucose “marks” beta cells resulting in preferential release of newly synthesize insulin. Science 1982; 218: 56–58 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U-M Protein Folding Diseases Initiative/International
- R01DK111174/GF/International
- R01 DK048280/DK/NIDDK NIH HHS/United States
- 17ZXMFSY00150/Tianjin Municipal Science and Technology Commission/International
- 81370895/National Natural Science Foundation of China/International
- R01 DK040949/DK/NIDDK NIH HHS/United States
- R24 DK110973/DK/NIDDK NIH HHS/United States
- R01DK113171/GF/International
- R01 DK113171/DK/NIDDK NIH HHS/United States
- R01DK48280/GF/International
- R01 DK111174/DK/NIDDK NIH HHS/United States
- 81620108004/National Natural Science Foundation of China/International
- P30 DK020572/DK/NIDDK NIH HHS/United States
- R24DK110973/GF/International
- R01 DK079233/DK/NIDDK NIH HHS/United States
- 81570699/National Natural Science Foundation of China/International
- T32 GM007250/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
