

Peptide and Protein Structure Prediction with a Simplified Continuum Solvent Model
- PMID: 30230838
- PMCID: PMC6741432
- DOI: 10.1021/acs.jpcb.8b07264
Peptide and Protein Structure Prediction with a Simplified Continuum Solvent Model
Abstract
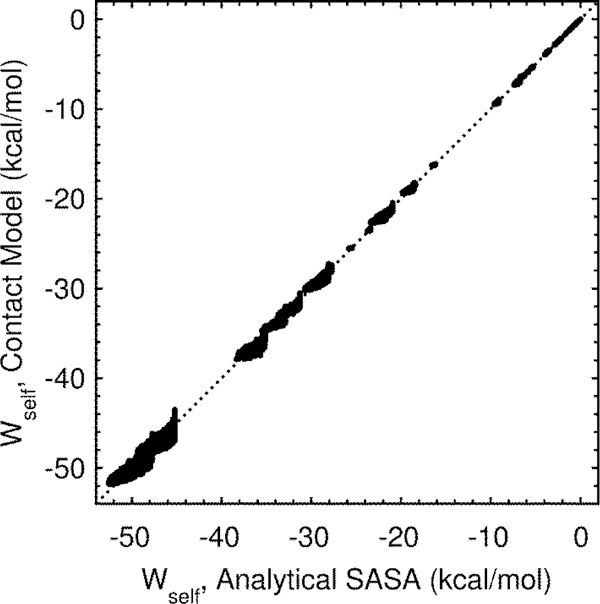
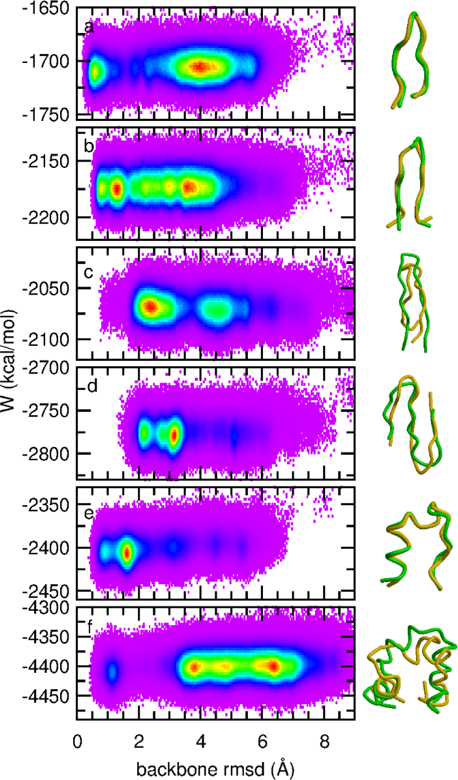
A continuum solvent model based on screened Coulomb potentials has been simplified and parametrized to sample native-like structures in replica-exchange simulations of each of six different peptides and miniproteins. Low-energy, native, and non-native structures were used to iteratively refine 11 parameter values. The centroid of the largest cluster of structures sampled in simulations initiated from an extended conformation represents the predicted structure. The main-chain rms deviation of this prediction from the experimental structure was 0.47 Å for the 12-residue Trp-zip2, 0.86 Å for the 14-residue MBH12, 2.53 Å for the 17-residue U(1-17)T9D, 2.03 Å for the 20-residue BS1, 1.08 Å for the 20-residue Trp-cage, and 3.64 Å for the 35-residue villin headpiece subdomain HP35. The centroid of the sixth largest cluster sampled for HP35 deviated by 0.91 Å. The CHARMM22/CMAP force field was used, with an additional ψ torsion term for residues other than glycine and proline. Six parameters govern the dielectric response of the continuum solvent, and four values of surface tension approximate nonpolar effects. An atom's self-energy and interaction energies are screened independently, each depending on whether the atom is part of a charged group, a neutral hydrogen-bonding main-chain group, or any other neutral group. The parameters inferred result in strong main-chain hydrogen bonds, consistent with the view that protein folding is dominated by the formation of these bonds. (1,2) Conformations of MBH12 and BS1 were excluded from the energy-function refinement, suggesting the parameters, referred to as SCP18, are transferable. An efficient estimate of solvent-accessible surface area is also described.
Figures

Similar articles
-
Long dynamics simulations of proteins using atomistic force fields and a continuum representation of solvent effects: calculation of structural and dynamic properties.Proteins. 2005 Aug 15;60(3):464-84. doi: 10.1002/prot.20470. Proteins. 2005. PMID: 15959866 Free PMC article.
-
Exploratory studies of ab initio protein structure prediction: multiple copy simulated annealing, AMBER energy functions, and a generalized born/solvent accessibility solvation model.Proteins. 2002 Jan 1;46(1):128-46. doi: 10.1002/prot.10020. Proteins. 2002. PMID: 11746709
-
Exploring peptide energy landscapes: a test of force fields and implicit solvent models.Proteins. 2004 Dec 1;57(4):665-77. doi: 10.1002/prot.20247. Proteins. 2004. PMID: 15390266
-
Molecular dynamics simulations of peptides and proteins with a continuum electrostatic model based on screened Coulomb potentials.Proteins. 2003 Apr 1;51(1):109-25. doi: 10.1002/prot.10330. Proteins. 2003. PMID: 12596268
-
Tri-peptide reference structures for the calculation of relative solvent accessible surface area in protein amino acid residues.Comput Biol Chem. 2015 Feb;54:33-43. doi: 10.1016/j.compbiolchem.2014.11.007. Epub 2014 Dec 3. Comput Biol Chem. 2015. PMID: 25544680
Cited by
-
Modulation of free energy landscapes as a strategy for the design of antimicrobial peptides.J Biol Phys. 2022 Jun;48(2):151-166. doi: 10.1007/s10867-022-09605-z. Epub 2022 Apr 14. J Biol Phys. 2022. PMID: 35419659 Free PMC article.
References
-
- Bolen DW; Rose GD Structure and Energetics of the Hydrogen-Bonded Backbone in Protein Folding. Annu. Rev. Biohchem 2008, 77 (1), 339–362. - PubMed
-
- Lindorff-Larsen K; Piana S; Dror RO; Shaw DE How Fast-Folding Proteins Fold. Science 2011, 334 (6055), 517–520. - PubMed
-
- Gilson MK; Davis ME; Luty BA; McCammon JA Computation of Electrostatic Forces on Solvated Molecules using the Poisson-Boltzmann Equation. J. Phys. Chem 1993, 97 (14), 3591–3600.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
