

Exploring transcription factors reveals crucial members and regulatory networks involved in different abiotic stresses in Brassica napus L
- PMID: 30231862
- PMCID: PMC6146658
- DOI: 10.1186/s12870-018-1417-z
Exploring transcription factors reveals crucial members and regulatory networks involved in different abiotic stresses in Brassica napus L
Abstract
Background: Brassica napus (B. napus) encompasses diverse transcription factors (TFs), but thorough identification and characterization of TF families, as well as their transcriptional responsiveness to multifarious stresses are still not clear.
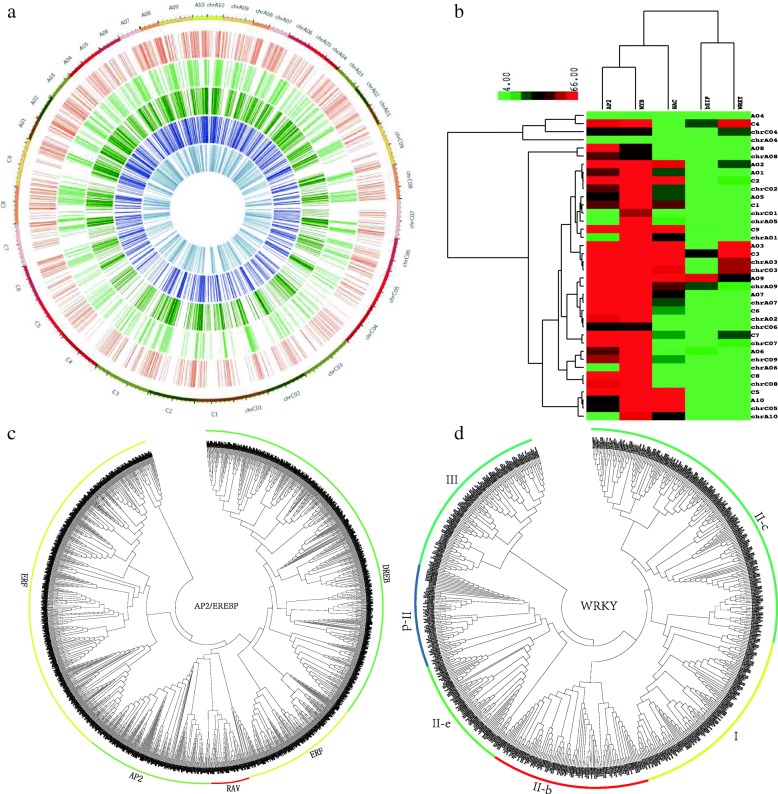
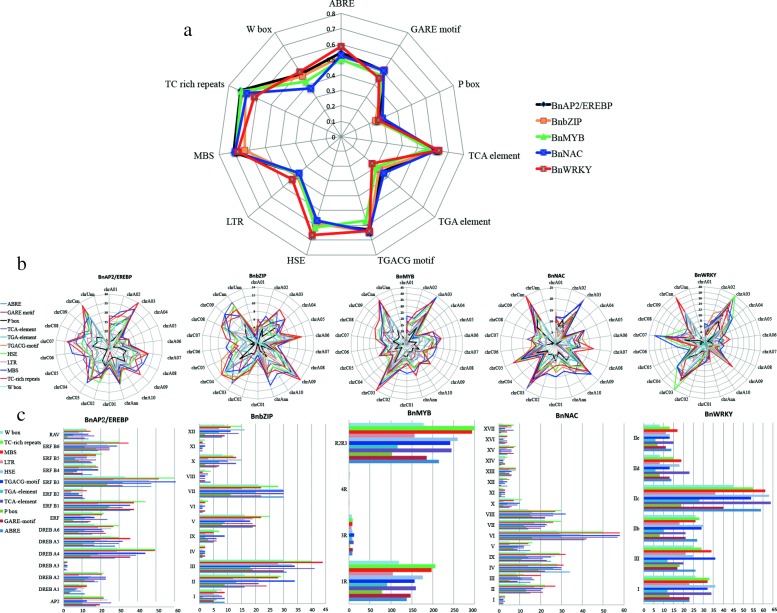
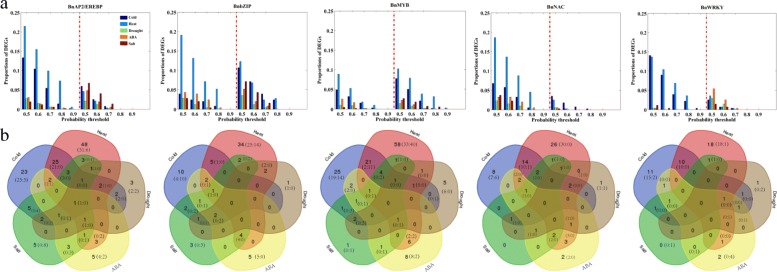
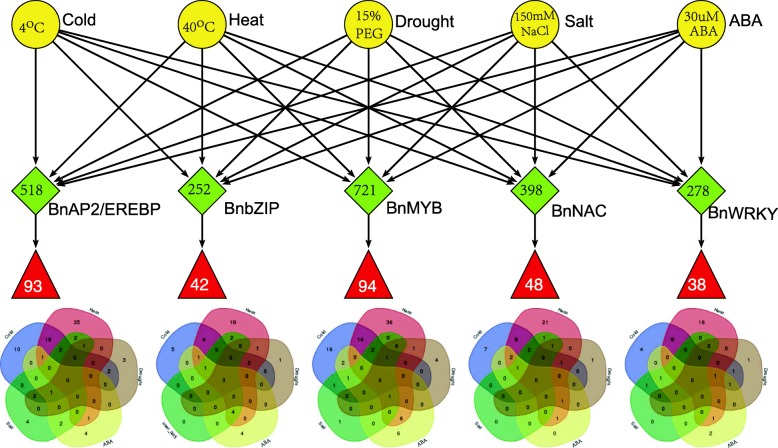
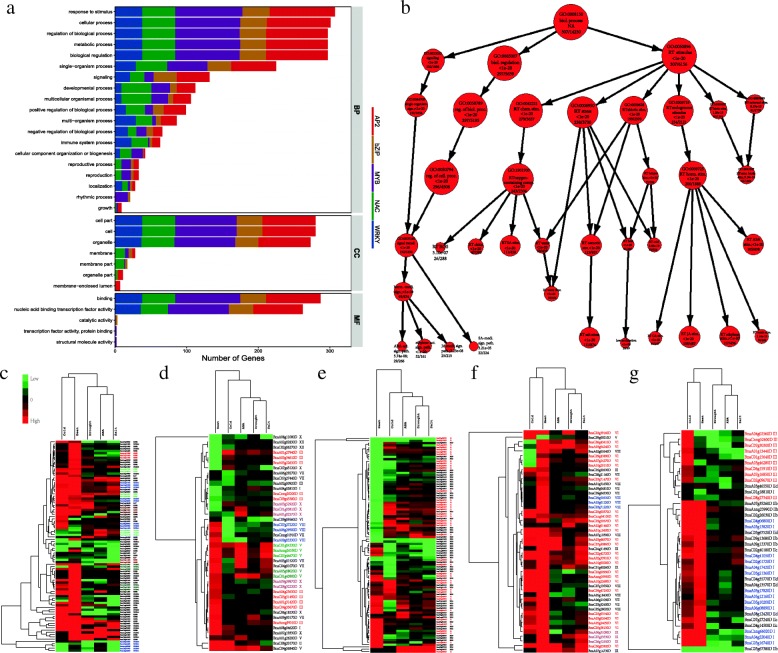
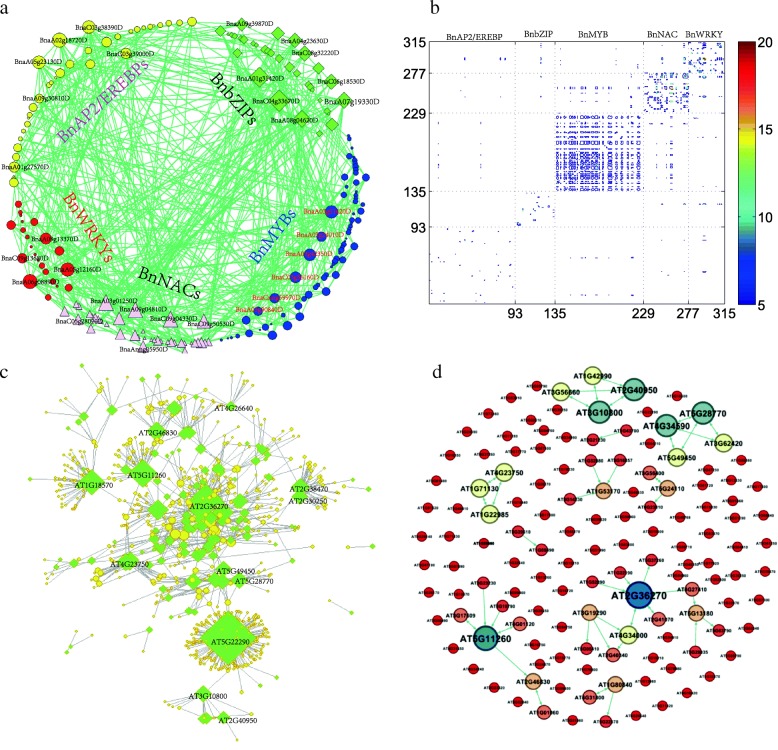
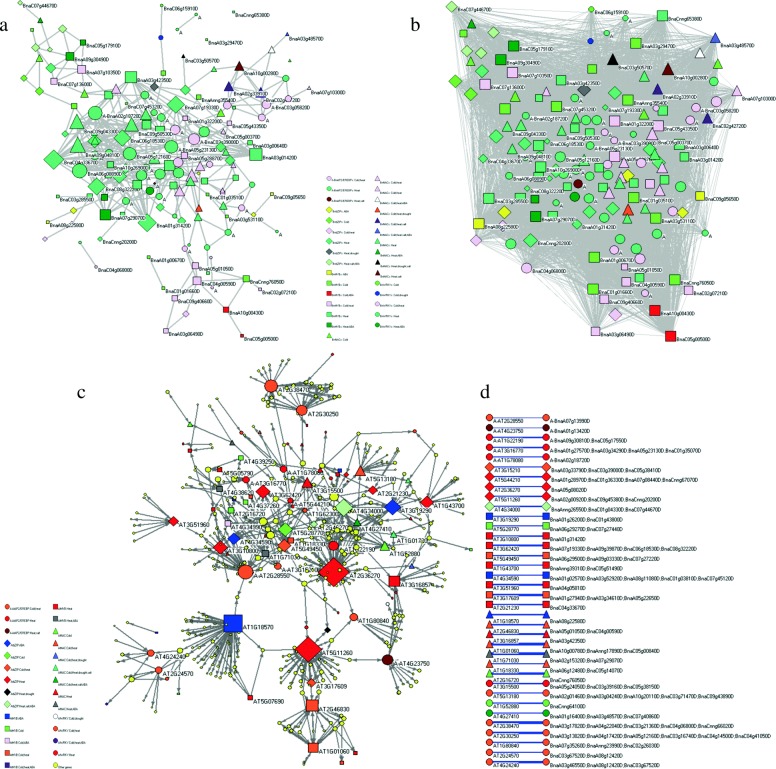
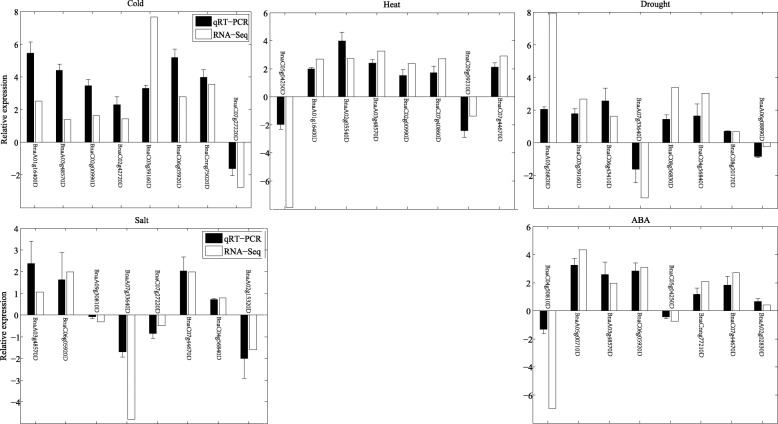
Results: Totally 2167 TFs belonging to five families were genome-widely identified in B. napus, including 518 BnAP2/EREBPs, 252 BnbZIPs, 721 BnMYBs, 398 BnNACs and 278 BnWRKYs, which contained some novel members in comparison with existing results. Sub-genome distributions of BnAP2/EREBPs and BnMYBs indicated that the two families might have suffered from duplication and divergence during evolution. Synteny analysis revealed strong co-linearity between B. napus and its two ancestors, although chromosomal rearrangements have occurred and 85 TFs were lost. About 7.6% and 9.4% TFs of the five families in B. napus were novel genes and conserved genes, which both showed preference on the C sub-genome. RNA-Seq revealed that more than 80% TFs were abiotic stress inducible and 315 crucial differentially expressed genes (DEGs) were screened out. Network analysis revealed that the 315 DEGs are highly co-expressed. The homologous gene network in A. thaliana revealed that a considerable amount of TFs could trigger the differential expression of targeted genes, resulting in a complex clustered network with clusters of genes responsible for targeted stress responsiveness.
Conclusions: We identified and characterized five TF families in B. napus. Some crucial members and regulatory networks involved in different abiotic stresses have been explored. The investigations deepen our understanding of TFs for stress tolerance in B. napus.
Keywords: Abiotic stress; Brassica napus; Gene regulatory network; RNA-Seq; Transcription factor.
Conflict of interest statement
Ethics approval and consent to participate
The plant material Zhongshuang 11 was provided by the Oil Crops Research Institute (OCRI), Chinese Academy of Agricultural Sciences (CAAS), which is a popularized planting varieties in large area and can be freely used for research purpose.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Riechmann JL, Meyerowitz EM. The AP2/EREBP family of plant transcription factors. Biol Chem. 1998;379:633–46. - PubMed
MeSH terms
Substances
Grants and funding
- 2016YFD0101900/National Key Research and Development Program of China
- 61773153/National Natural Science Foundation of China
- 31671728/National Natural Science Foundation of China
- 17A120002/The Key Scientific Research Projects in Colleges and Universities of Henan
- 151100111200/Major science and technology special projects of Henan Province
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
