

Platelet heterogeneity in activation-induced glycoprotein shedding: functional effects
- PMID: 30232085
- PMCID: PMC6156892
- DOI: 10.1182/bloodadvances.2017011544
Platelet heterogeneity in activation-induced glycoprotein shedding: functional effects
Abstract
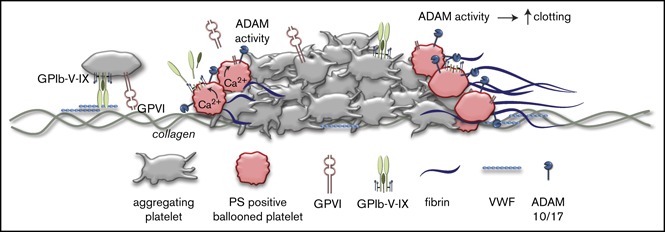
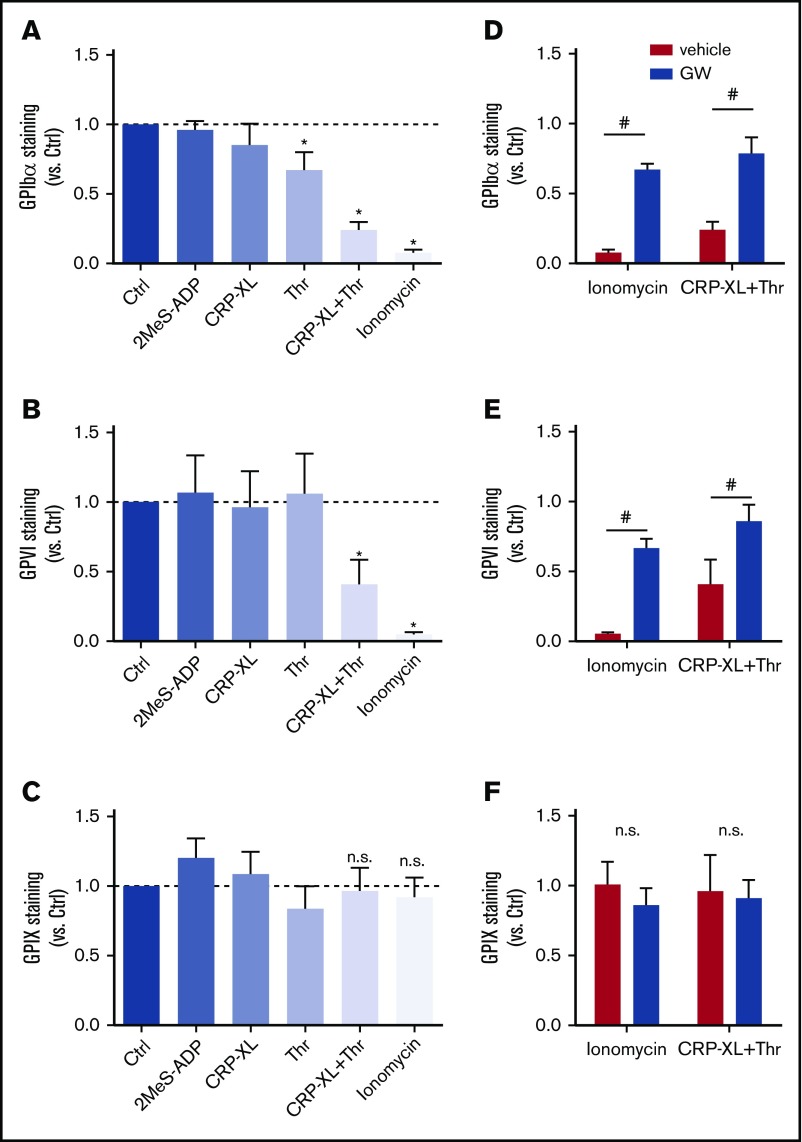
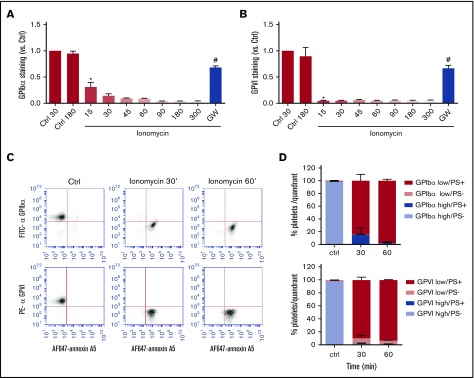
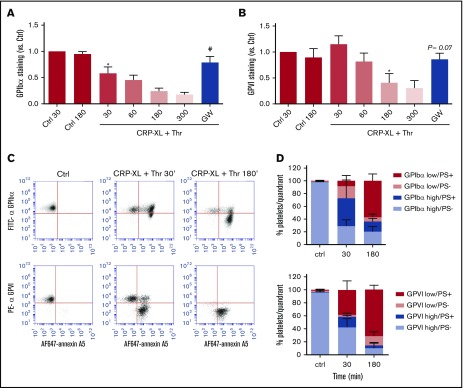
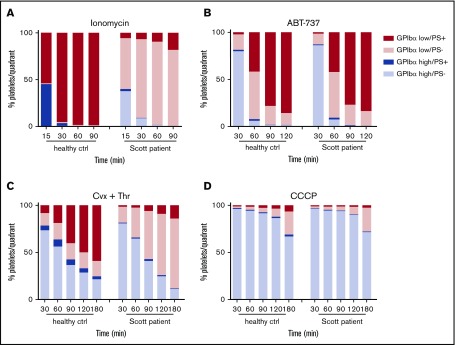
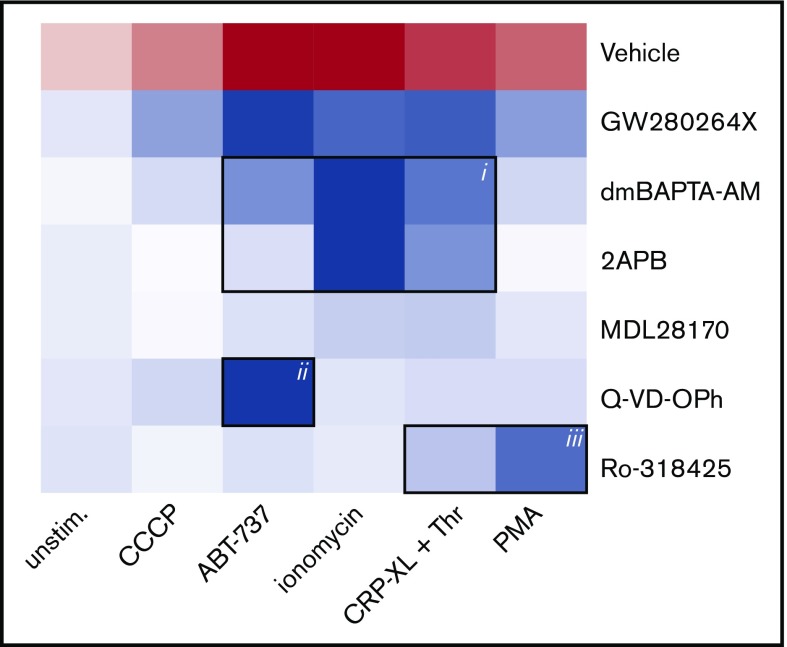
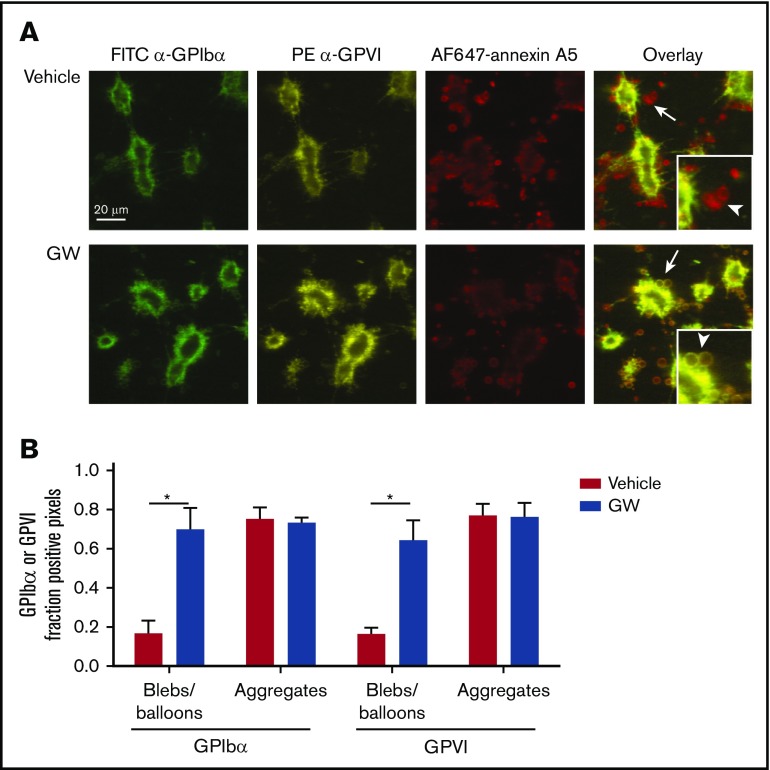
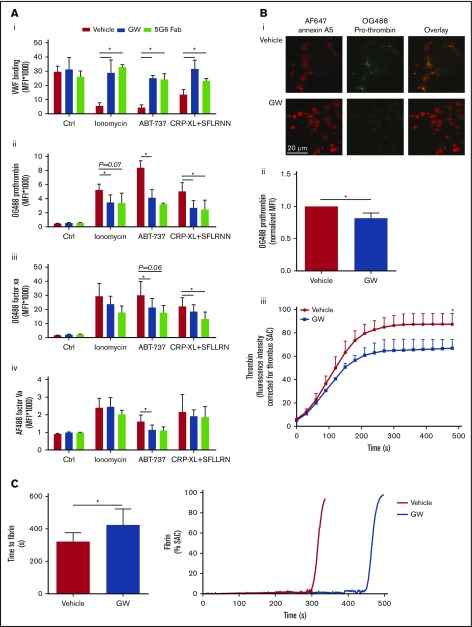
The platelet receptors glycoprotein Ibα (GPIbα) and GPVI are known to be cleaved by members of a disintegrin and metalloprotease (ADAM) family (ADAM10 and ADAM17), but the mechanisms and consequences of this shedding are not well understood. Our results revealed that (1) glycoprotein shedding is confined to distinct platelet populations showing near-complete shedding, (2) the heterogeneity between (non)shed platelets is independent of agonist type but coincides with exposure of phosphatidylserine (PS), and (3) distinct pathways of shedding are induced by elevated Ca2+, low Ca2+ protein kinase C (PKC), or apoptotic activation. Furthermore, we found that receptor shedding reduces binding of von Willebrand factor, enhances binding of coagulation factors, and augments fibrin formation. In response to Ca2+-increasing agents, shedding of GPIbα was abolished by ADAM10/17 inhibition but not by blockage of calpain. Stimulation of PKC induced shedding of only GPIbα, which was annulled by kinase inhibition. The proapoptotic agent ABT-737 induced shedding, which was caspase dependent. In Scott syndrome platelets that are deficient in Ca2+-dependent PS exposure, shedding occurred normally, indicating that PS exposure is not a prerequisite for ADAM activity. In whole-blood thrombus formation, ADAM-dependent glycoprotein shedding enhanced thrombin generation and fibrin formation. Together, these findings indicate that 2 major activation pathways can evoke ADAM-mediated glycoprotein shedding in distinct platelet populations and that shedding modulates platelet function from less adhesive to more procoagulant.
© 2018 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Figures
References
-
- Versteeg HH, Heemskerk JW, Levi M, Reitsma PH. New fundamentals in hemostasis. Physiol Rev. 2013;93(1):327-358. - PubMed
-
- Berndt MC, Metharom P, Andrews RK. Primary haemostasis: newer insights. Haemophilia. 2014;20(suppl 4):15-22. - PubMed
-
- Swieringa F, Kuijpers MJE, Heemskerk JW, van der Meijden PE. Targeting platelet receptor function in thrombus formation: the risk of bleeding. Blood Rev. 2014;28(1):9-21. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
