

Redundant Late Domain Functions of Tandem VP2 YPX3L Motifs in Nonlytic Cellular Egress of Quasi-enveloped Hepatitis A Virus
- PMID: 30232181
- PMCID: PMC6232465
- DOI: 10.1128/JVI.01308-18
Redundant Late Domain Functions of Tandem VP2 YPX3L Motifs in Nonlytic Cellular Egress of Quasi-enveloped Hepatitis A Virus
Abstract
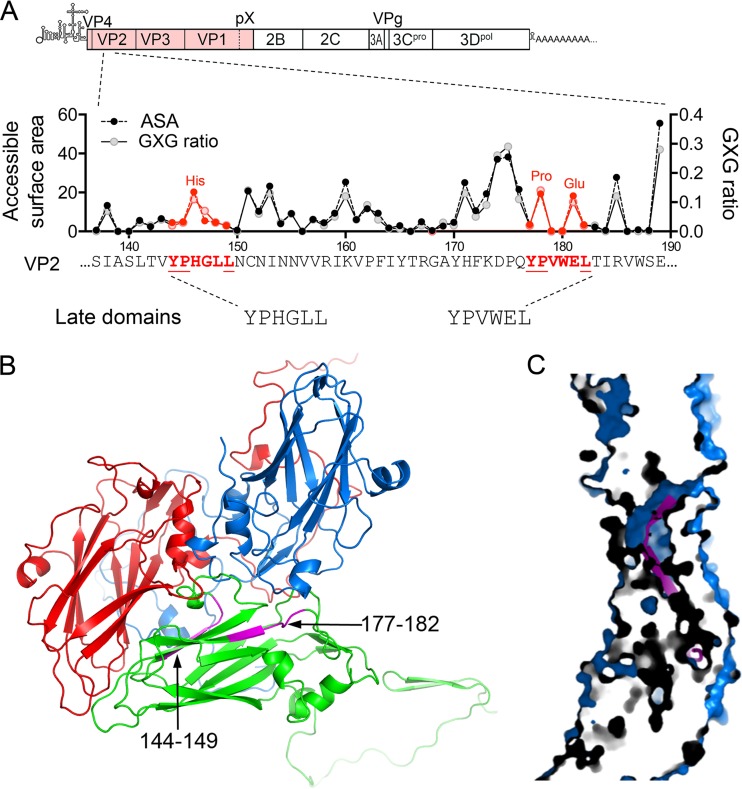
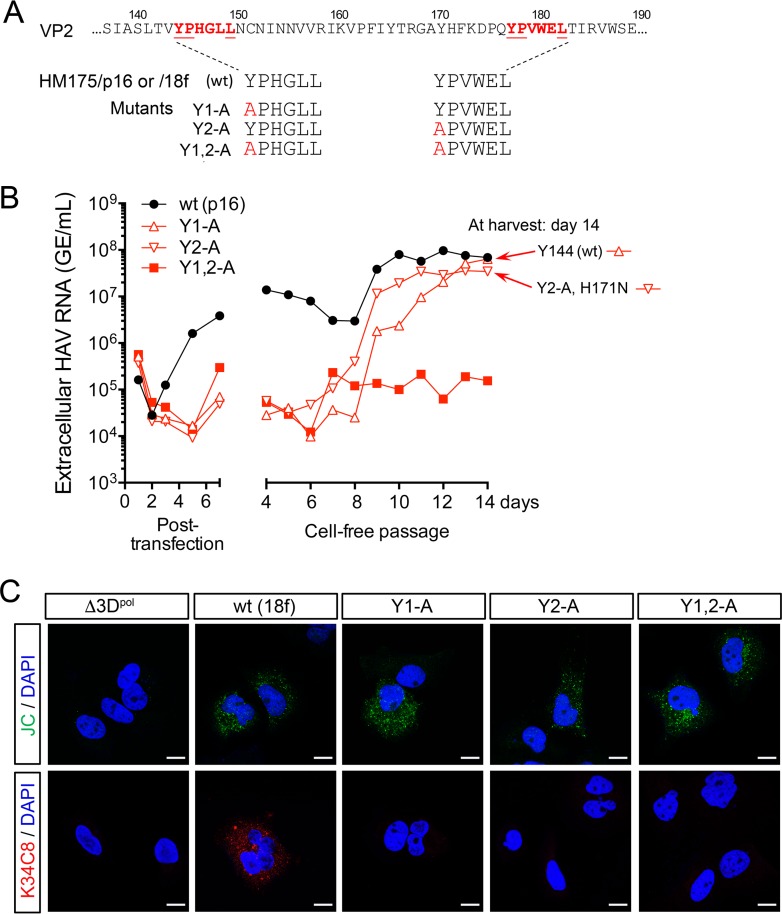
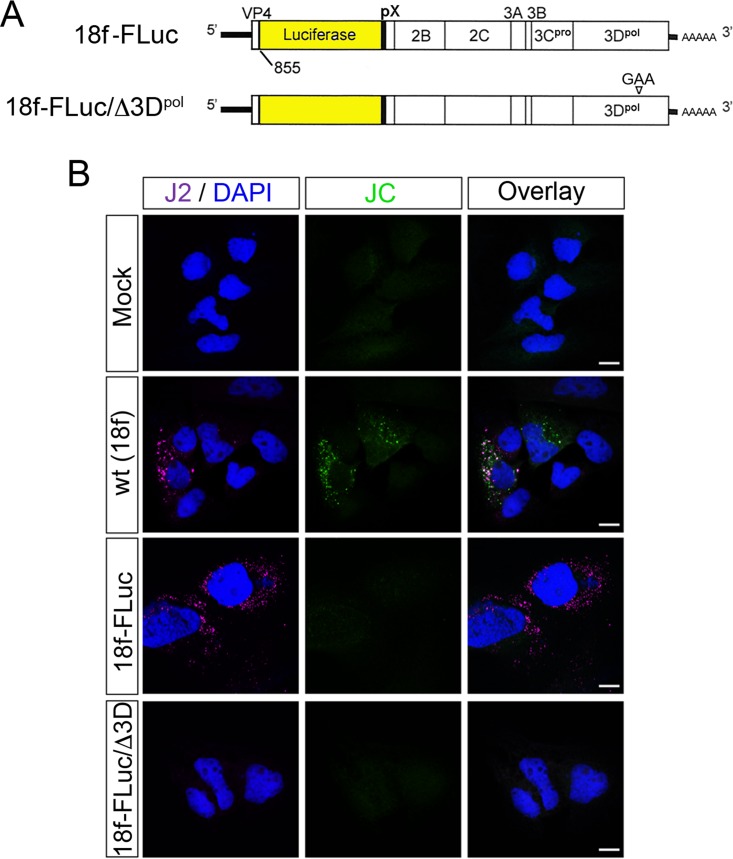
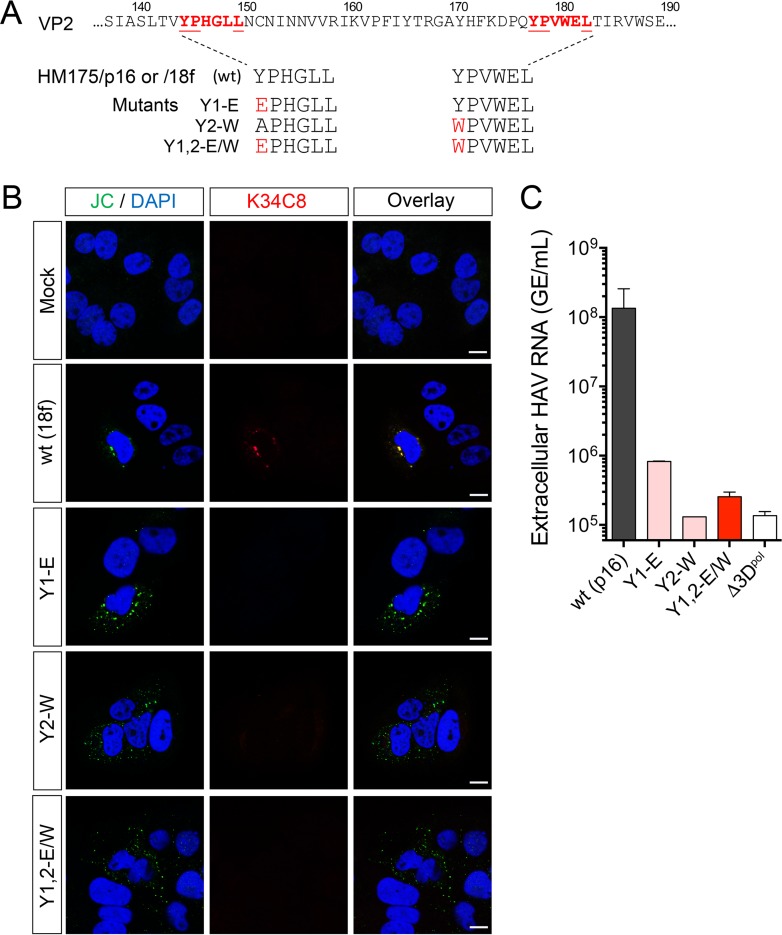
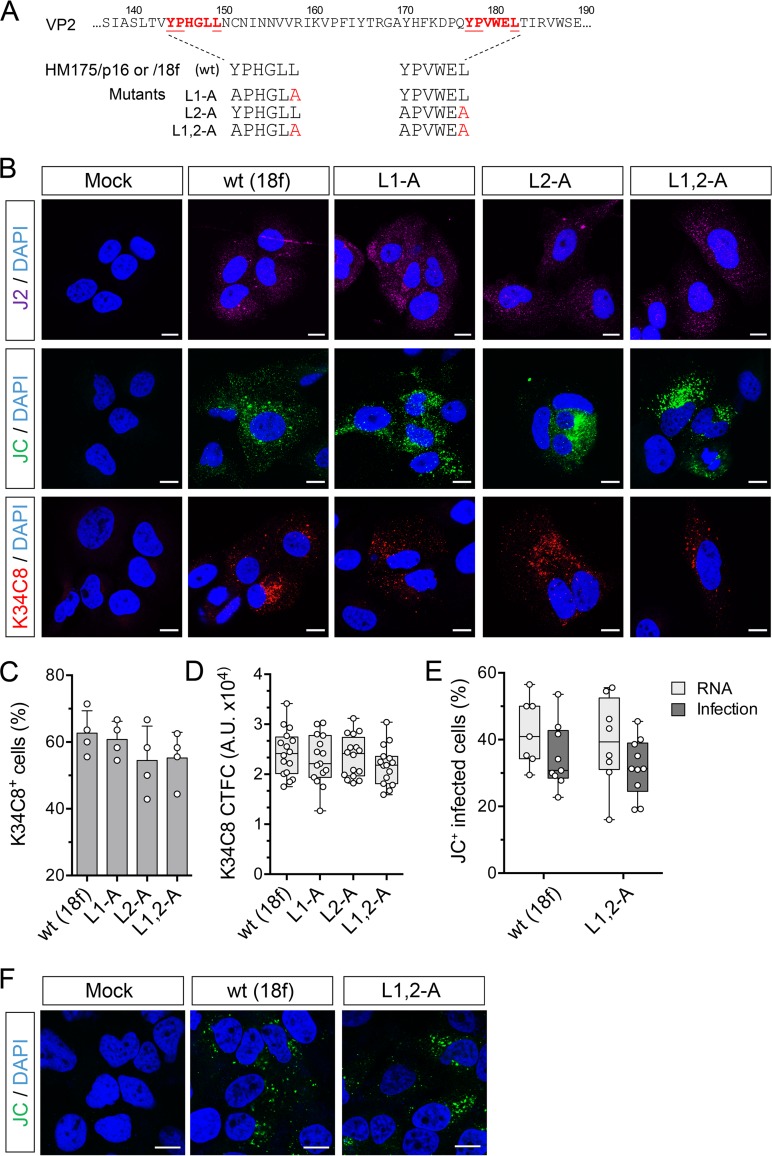
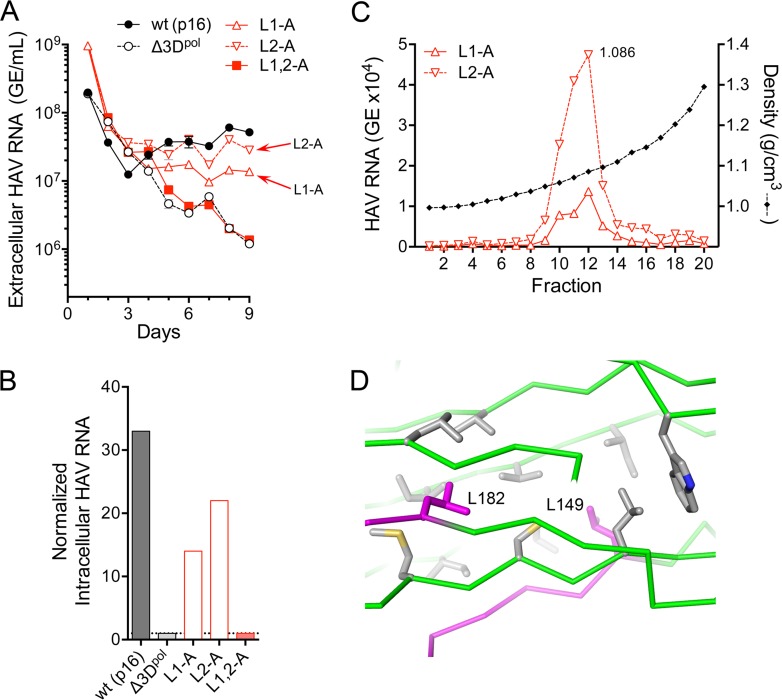
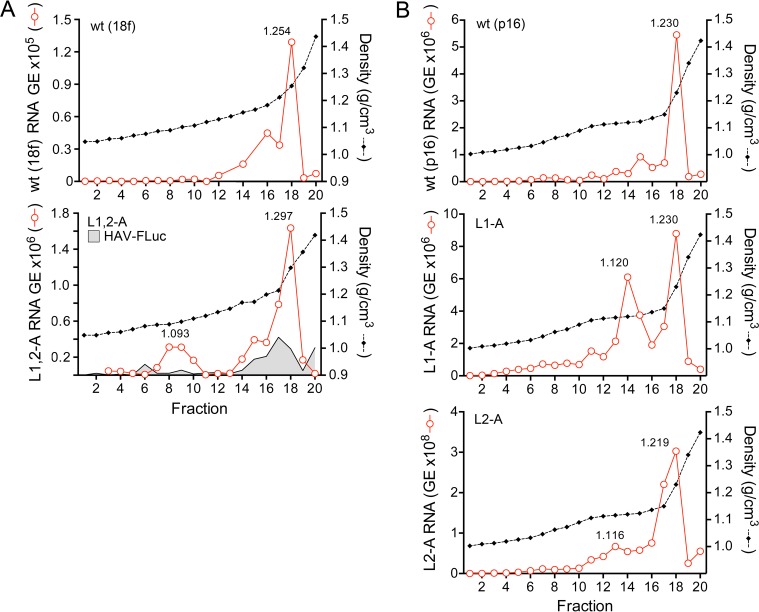
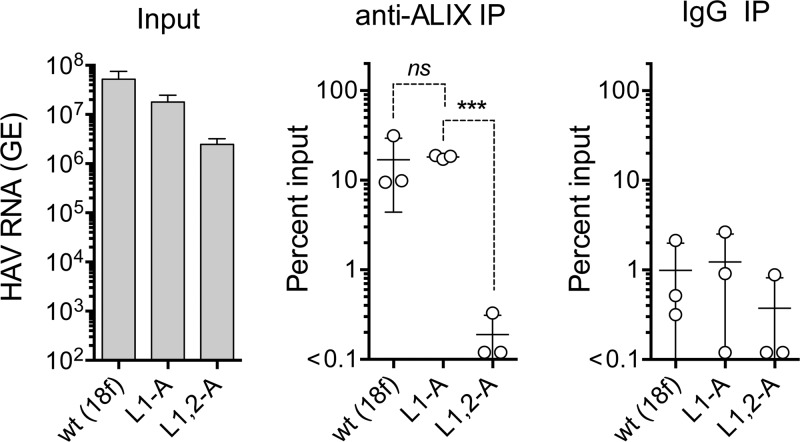
The quasi-envelopment of hepatitis A virus (HAV) capsids in exosome-like virions (eHAV) is an important but incompletely understood aspect of the hepatovirus life cycle. This process is driven by recruitment of newly assembled capsids to endosomal vesicles into which they bud to form multivesicular bodies with intraluminal vesicles that are later released at the plasma membrane as eHAV. The endosomal sorting complexes required for transport (ESCRT) are key to this process, as is the ESCRT-III-associated protein, ALIX, which also contributes to membrane budding of conventional enveloped viruses. YPX1or3L late domains in the structural proteins of these viruses mediate interactions with ALIX, and two such domains exist in the HAV VP2 capsid protein. Mutational studies of these domains are confounded by the fact that the Tyr residues (important for interactions of YPX1or3L peptides with ALIX) are required for efficient capsid assembly. However, single Leu-to-Ala substitutions within either VP2 YPX3L motif (L1-A and L2-A mutants) were well tolerated, albeit associated with significantly reduced eHAV release. In contrast, simultaneous substitutions in both motifs (L1,2-A) eliminated virus release but did not inhibit assembly of infectious intracellular particles. Immunoprecipitation experiments suggested that the loss of eHAV release was associated with a loss of ALIX recruitment. Collectively, these data indicate that HAV YPX3L motifs function as redundant late domains during quasi-envelopment and viral release. Since these motifs present little solvent-accessible area in the crystal structure of the naked extracellular capsid, the capsid structure may be substantially different during quasi-envelopment.IMPORTANCE Nonlytic release of hepatitis A virus (HAV) as exosome-like quasi-enveloped virions is a unique but incompletely understood aspect of the hepatovirus life cycle. Several lines of evidence indicate that the host protein ALIX is essential for this process. Tandem YPX3L "late domains" in the VP2 capsid protein could be sites of interaction with ALIX, but they are not accessible on the surface of an X-ray model of the extracellular capsid, raising doubts about this putative late domain function. Here, we describe YPX3L domain mutants that assemble capsids normally but fail to bind ALIX and be secreted as quasi-enveloped eHAV. Our data support late domain function for the VP2 YPX3L motifs and raise questions about the structure of the HAV capsid prior to and following quasi-envelopment.
Keywords: ESCRT; exosome; picornavirus; quasi-envelope.
Copyright © 2018 American Society for Microbiology.
Figures
References
-
- Lanford RE, Feng Z, Chavez D, Guerra B, Brasky KM, Zhou Y, Yamane D, Perelson AS, Walker CM, Lemon SM. 2011. Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proc Natl Acad Sci U S A 108:11223–11228. doi: 10.1073/pnas.1101939108. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
