

T5 Exonuclease Hydrolysis of Hepatitis B Virus Replicative Intermediates Allows Reliable Quantification and Fast Drug Efficacy Testing of Covalently Closed Circular DNA by PCR
- PMID: 30232183
- PMCID: PMC6232461
- DOI: 10.1128/JVI.01117-18
T5 Exonuclease Hydrolysis of Hepatitis B Virus Replicative Intermediates Allows Reliable Quantification and Fast Drug Efficacy Testing of Covalently Closed Circular DNA by PCR
Abstract
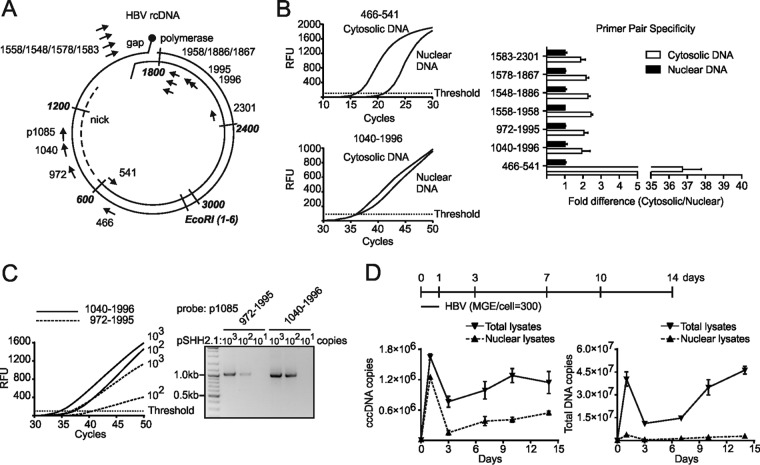
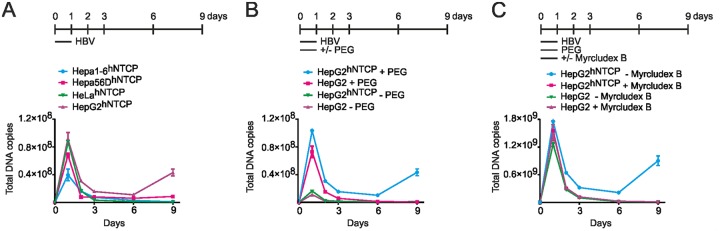
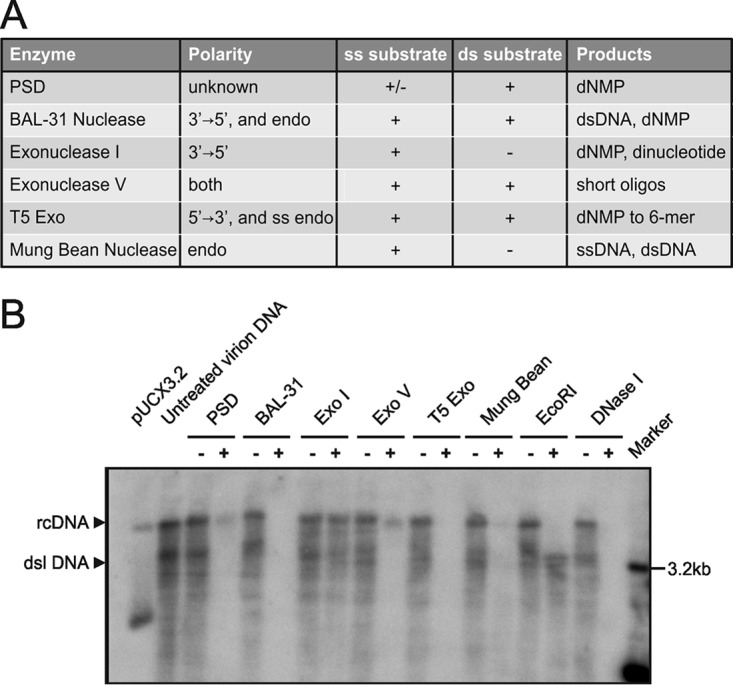
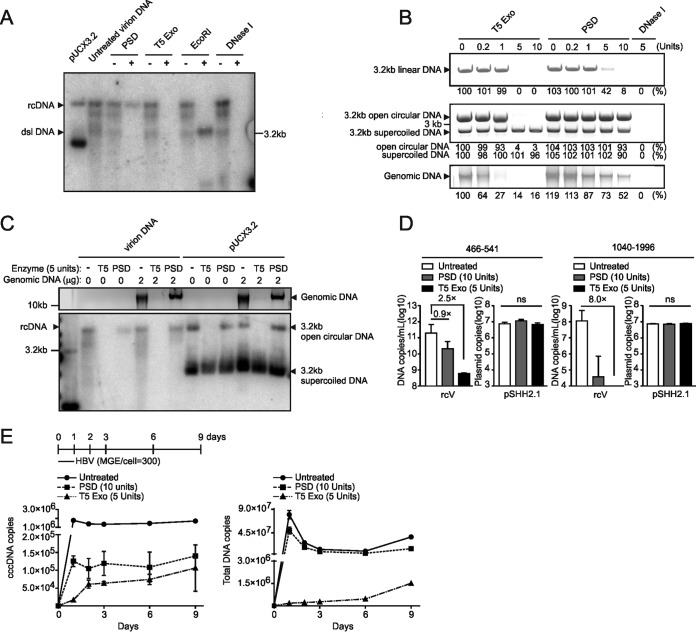
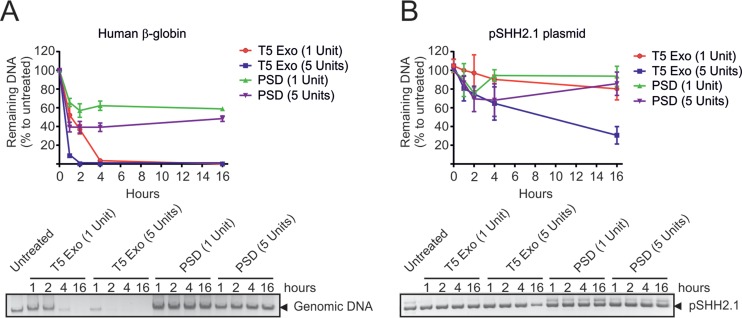
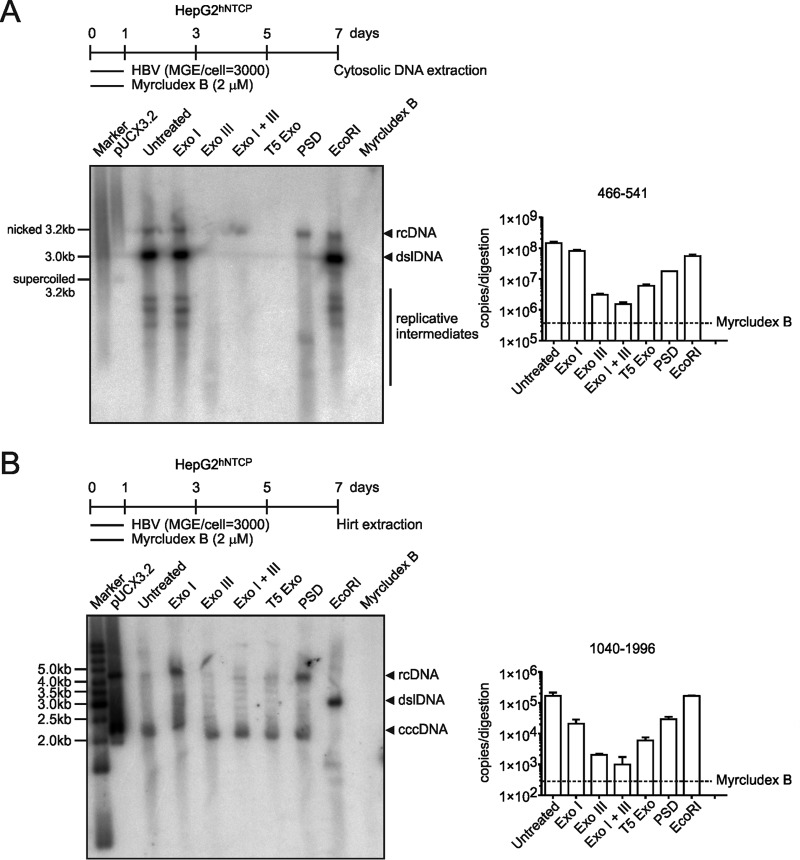
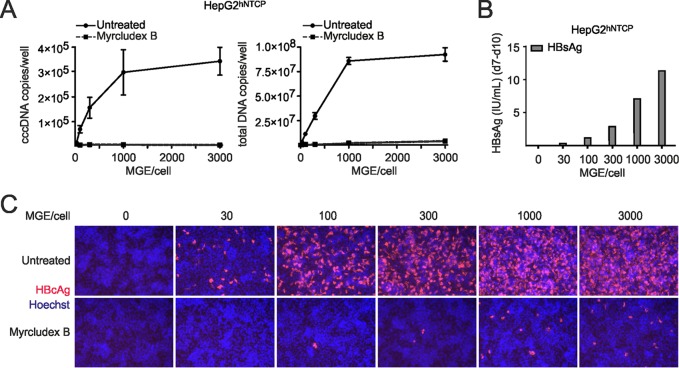
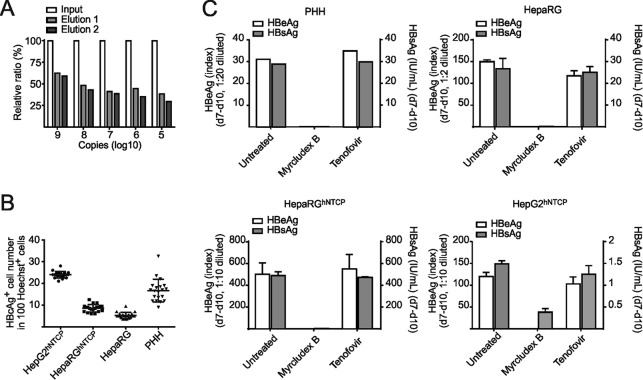
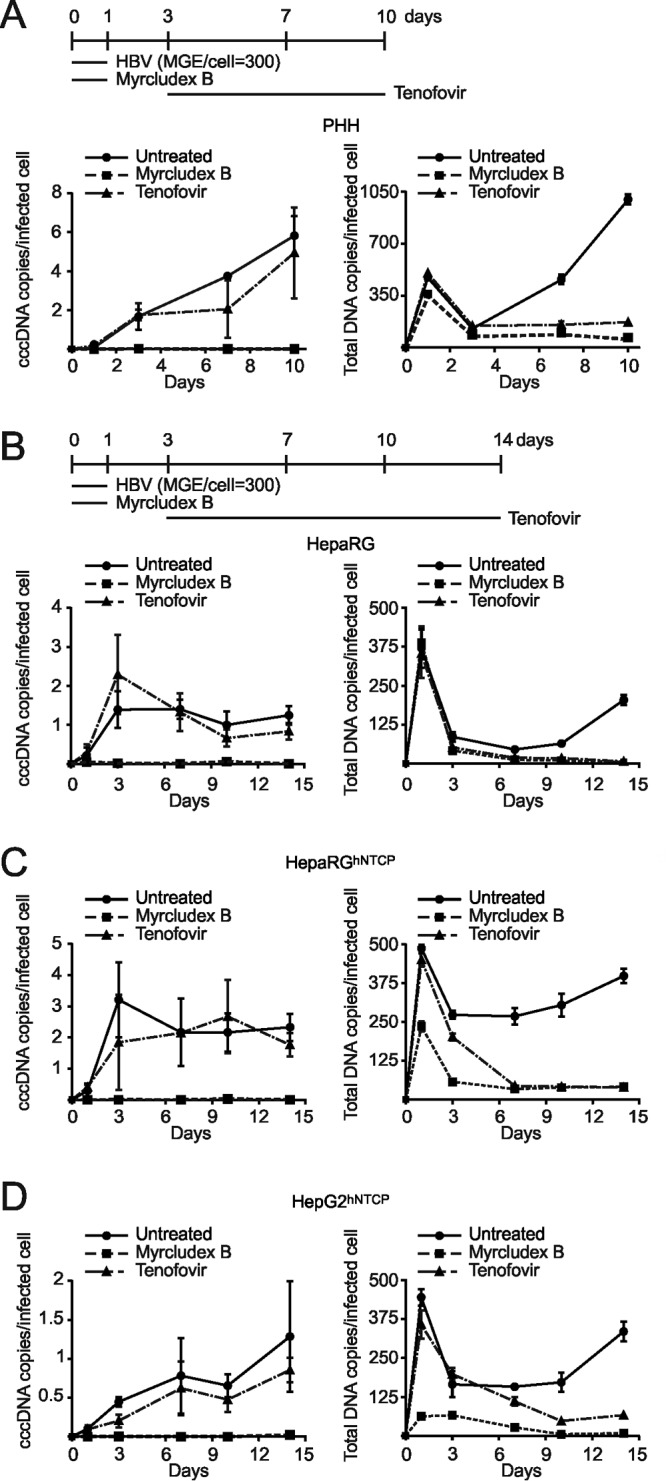
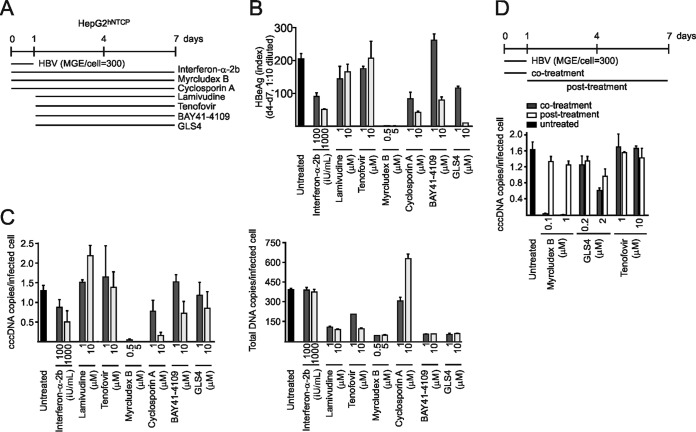
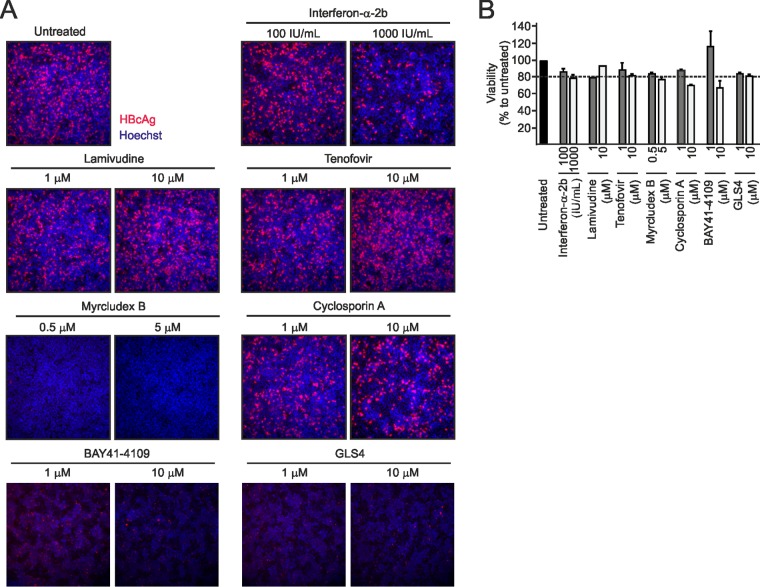
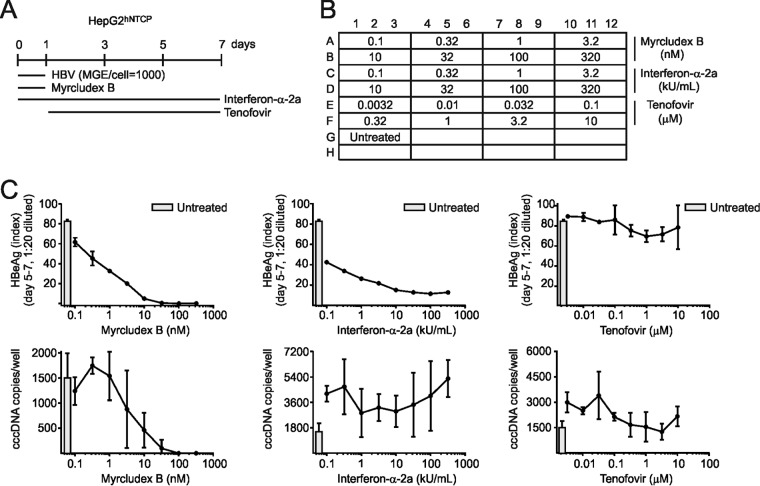
Chronic infection with the human hepatitis B virus (HBV) is a major health problem. Virus persistence requires the establishment and maintenance of covalently closed circular DNA (cccDNA), the episomal virus template in the nucleus of infected hepatocytes. Compared to replicative DNA intermediates (relaxed circular DNA [rcDNA]), copy numbers of cccDNA in infected hepatocytes are low. Accordingly, accurate analyses of cccDNA require enrichment of nuclear fractions and Southern blotting or selective quantitative PCR (qPCR) methods allowing discrimination of cccDNA and rcDNA. In this report, we analyzed cccDNA-specific primer pairs for their ability to amplify cccDNA selectively. Using mixtures of defined forms of HBV and genomic DNA, we determined the potential of different nucleases for targeted digestion of the open/relaxed circular DNA forms in the absence and presence of genomic DNA without affecting cccDNA. We found that the combination of T5 exonuclease with a primer set amplifying an approximately 1-kb fragment permits reliable quantification of cccDNA without the requirement of prior nucleus enrichment or Hirt extraction. We tested this method in four different in vitro infection systems and quantified cccDNA copy numbers at increasing multiplicities of inoculated genome equivalents. We further analyzed the kinetics of cccDNA formation and the effect of drugs (interferon, entry inhibitors, and capsid inhibitors) on cccDNA. Our method allows reliable cccDNA quantification at early stages of infection in the presence of a high excess of input virus and replicative intermediates and is thereby suitable for drug screening and investigation of cccDNA formation and maintenance.IMPORTANCE cccDNA elimination is a major goal in future curative regimens for chronic HBV patients. However, PCR-based assays for cccDNA quantification show a principally constrained specificity when high levels of input virus or replicative intermediates are present. Here, we characterized T5 exonuclease as a suitable enzyme for medium-throughput in vitro assays that preserves cccDNA but efficiently removes rcDNA prior to PCR-based quantification. We compared T5 exonuclease with the previously described exonuclease III and showed that both nucleases are suitable for reliable quantification of cccDNA by PCR. We substantiated the applicability of our method through examination of early cccDNA formation and stable accumulation in several in vitro infection models and analyzed cccDNA stability after administration of anti-HBV drugs. Our results support the use of T5 exonuclease for fast and convenient rcDNA removal, especially for early cccDNA quantification and rapid drug testing in in vitro studies.
Keywords: Myrcludex B; T5 exonuclease; cccDNA; covalently closed circular DNA; hepatitis B virus.
Copyright © 2018 American Society for Microbiology.
Figures
Similar articles
-
Quantification of the hepatitis B virus cccDNA: evidence-based guidelines for monitoring the key obstacle of HBV cure.Gut. 2023 May;72(5):972-983. doi: 10.1136/gutjnl-2022-328380. Epub 2023 Jan 27. Gut. 2023. PMID: 36707234 Free PMC article.
-
Characterization of the Termini of Cytoplasmic Hepatitis B Virus Deproteinated Relaxed Circular DNA.J Virol. 2020 Dec 9;95(1):e00922-20. doi: 10.1128/JVI.00922-20. Print 2020 Dec 9. J Virol. 2020. PMID: 33055252 Free PMC article.
-
Untying relaxed circular DNA of hepatitis B virus by polymerase reaction provides a new option for accurate quantification and visualization of covalently closed circular DNA.J Gen Virol. 2022 Feb;103(2). doi: 10.1099/jgv.0.001591. J Gen Virol. 2022. PMID: 35130138
-
Hepatitis B Virus Capsid: The Core in Productive Entry and Covalently Closed Circular DNA Formation.Viruses. 2023 Feb 28;15(3):642. doi: 10.3390/v15030642. Viruses. 2023. PMID: 36992351 Free PMC article. Review.
-
Metabolism and function of hepatitis B virus cccDNA: Implications for the development of cccDNA-targeting antiviral therapeutics.Antiviral Res. 2015 Oct;122:91-100. doi: 10.1016/j.antiviral.2015.08.005. Epub 2015 Aug 10. Antiviral Res. 2015. PMID: 26272257 Free PMC article. Review.
Cited by
-
CRISPR/Cas13-assisted hepatitis B virus covalently closed circular DNA detection.Hepatol Int. 2022 Apr;16(2):306-315. doi: 10.1007/s12072-022-10311-0. Epub 2022 Mar 17. Hepatol Int. 2022. PMID: 35298777 Free PMC article.
-
A nucleosome switch primes Hepatitis B Virus infection.bioRxiv [Preprint]. 2024 Jun 12:2023.03.03.531011. doi: 10.1101/2023.03.03.531011. bioRxiv. 2024. Update in: Cell. 2025 Apr 17;188(8):2111-2126.e21. doi: 10.1016/j.cell.2025.01.033. PMID: 38915612 Free PMC article. Updated. Preprint.
-
Liver mechanosignaling as a natural anti-hepatitis B virus mechanism.Nat Commun. 2024 Sep 27;15(1):8375. doi: 10.1038/s41467-024-52718-3. Nat Commun. 2024. PMID: 39333106 Free PMC article.
-
Host cell-dependent late entry step as determinant of hepatitis B virus infection.PLoS Pathog. 2022 Jun 17;18(6):e1010633. doi: 10.1371/journal.ppat.1010633. eCollection 2022 Jun. PLoS Pathog. 2022. PMID: 35714170 Free PMC article.
-
Transient and tunable CRISPRa regulation of APOBEC/AID genes for targeting hepatitis B virus.Mol Ther Nucleic Acids. 2023 Apr 20;32:478-493. doi: 10.1016/j.omtn.2023.04.016. eCollection 2023 Jun 13. Mol Ther Nucleic Acids. 2023. PMID: 37187708 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
