

Young genes have distinct gene structure, epigenetic profiles, and transcriptional regulation
- PMID: 30232198
- PMCID: PMC6211652
- DOI: 10.1101/gr.234872.118
Young genes have distinct gene structure, epigenetic profiles, and transcriptional regulation
Abstract
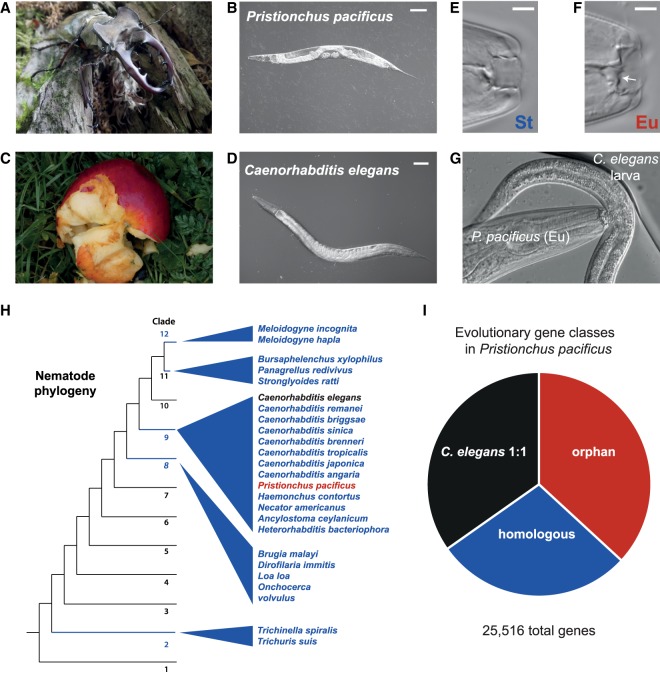
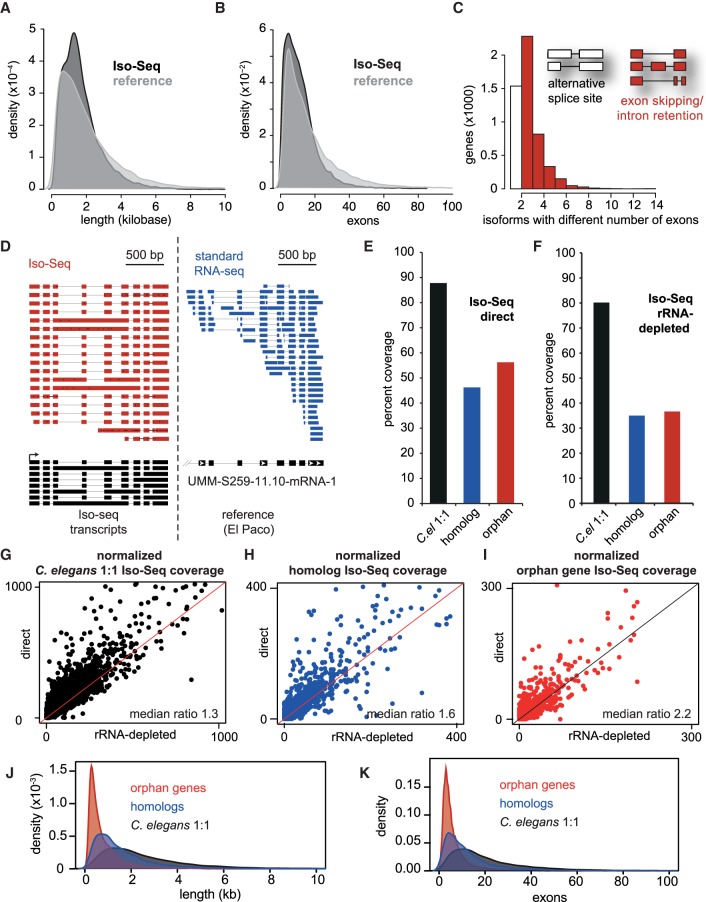
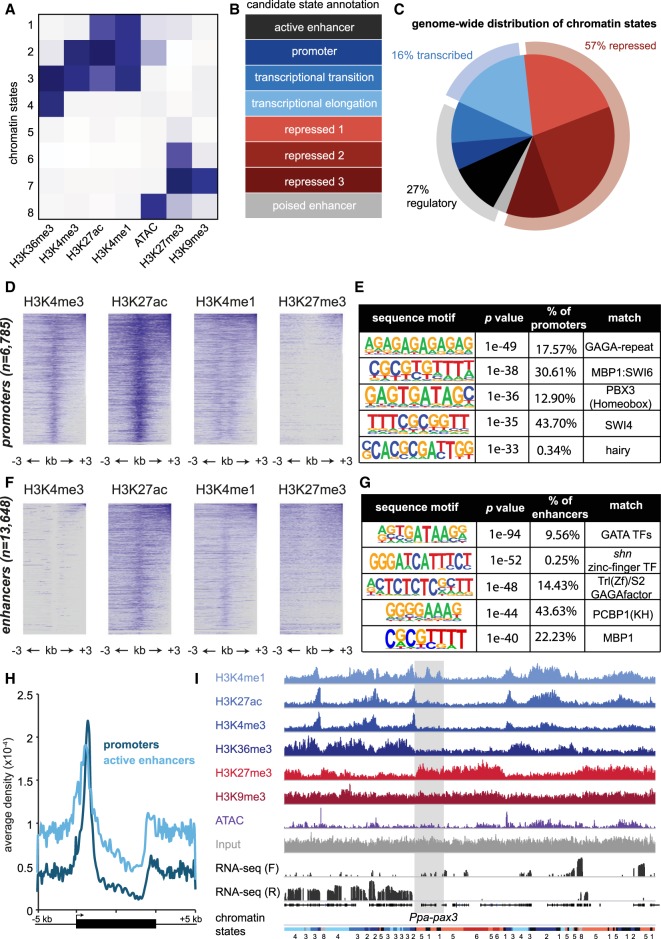
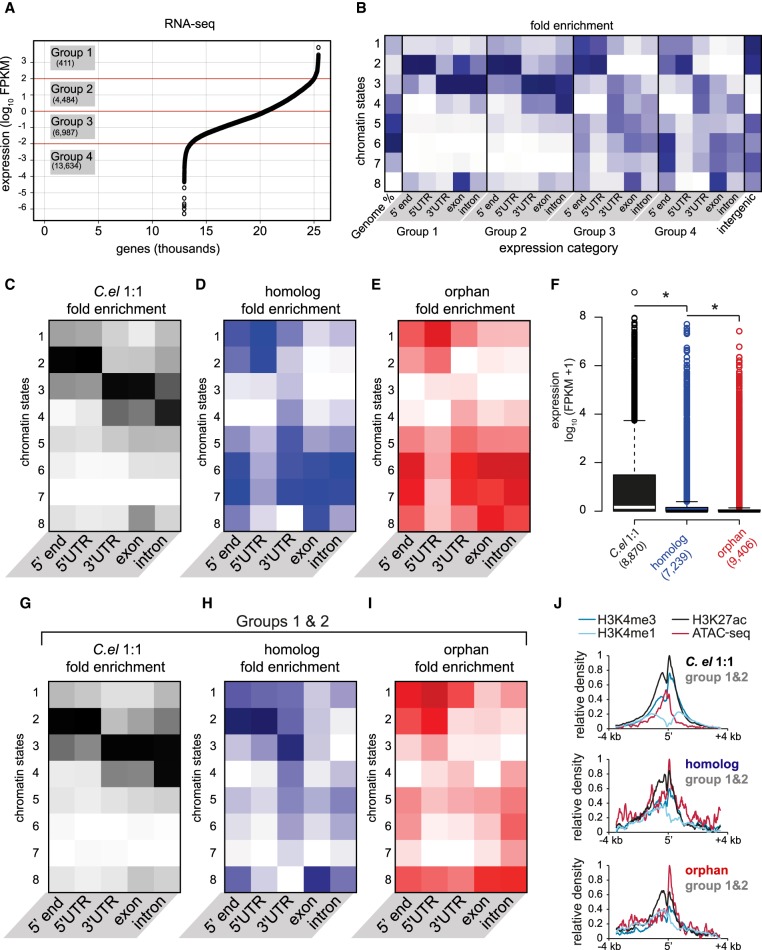
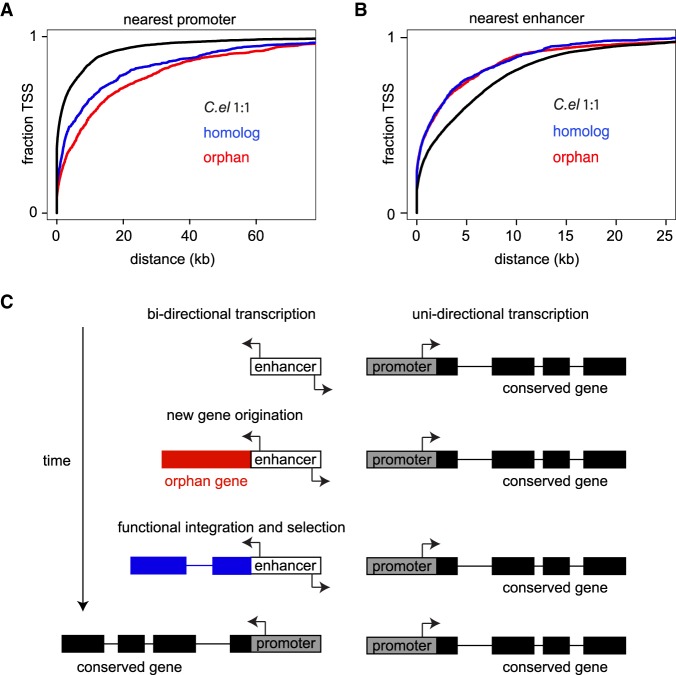
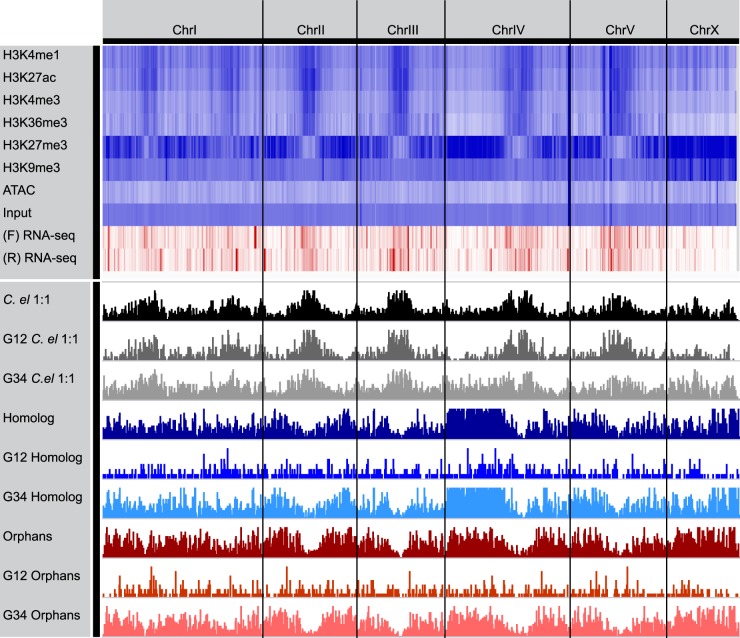
Species-specific, new, or "orphan" genes account for 10%-30% of eukaryotic genomes. Although initially considered to have limited function, an increasing number of orphan genes have been shown to provide important phenotypic innovation. How new genes acquire regulatory sequences for proper temporal and spatial expression is unknown. Orphan gene regulation may rely in part on origination in open chromatin adjacent to preexisting promoters, although this has not yet been assessed by genome-wide analysis of chromatin states. Here, we combine taxon-rich nematode phylogenies with Iso-Seq, RNA-seq, ChIP-seq, and ATAC-seq to identify the gene structure and epigenetic signature of orphan genes in the satellite model nematode Pristionchus pacificus Consistent with previous findings, we find young genes are shorter, contain fewer exons, and are on average less strongly expressed than older genes. However, the subset of orphan genes that are expressed exhibit distinct chromatin states from similarly expressed conserved genes. Orphan gene transcription is determined by a lack of repressive histone modifications, confirming long-held hypotheses that open chromatin is important for new gene formation. Yet orphan gene start sites more closely resemble enhancers defined by H3K4me1, H3K27ac, and ATAC-seq peaks, in contrast to conserved genes that exhibit traditional promoters defined by H3K4me3 and H3K27ac. Although the majority of orphan genes are located on chromosome arms that contain high recombination rates and repressive histone marks, strongly expressed orphan genes are more randomly distributed. Our results support a model of new gene origination by rare integration into open chromatin near enhancers.
© 2018 Werner et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Andersson R, Sandelin A, Danko CG. 2015. A unified architecture of transcriptional regulatory elements. Trends Genet 31: 426–433. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources