

Impairments in Oxidative Glucose Metabolism in Epilepsy and Metabolic Treatments Thereof
- PMID: 30233320
- PMCID: PMC6127311
- DOI: 10.3389/fncel.2018.00274
Impairments in Oxidative Glucose Metabolism in Epilepsy and Metabolic Treatments Thereof
Abstract
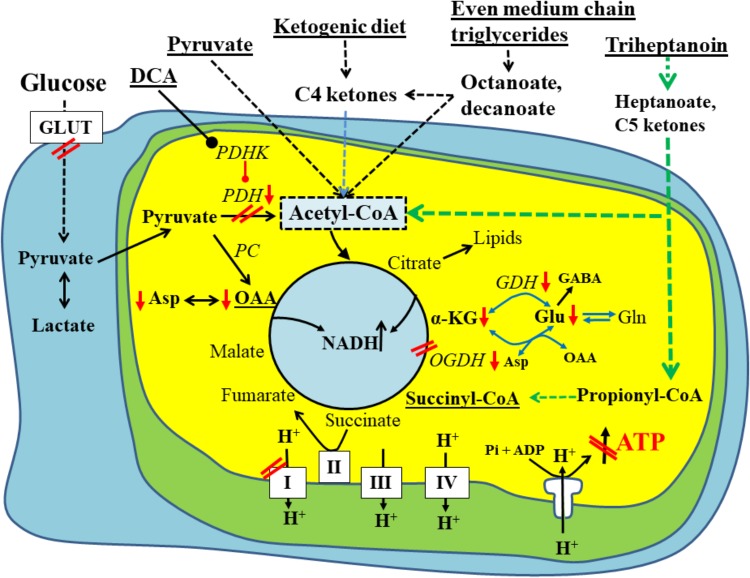
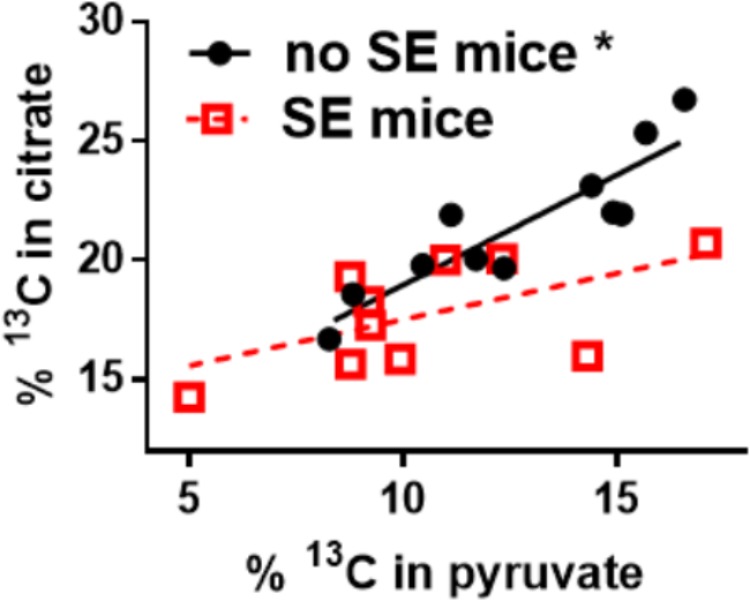
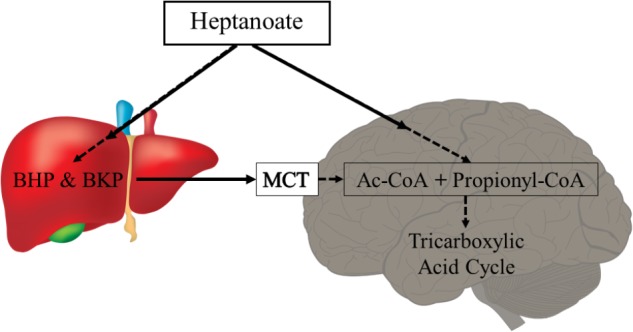
There is mounting evidence that oxidative glucose metabolism is impaired in epilepsy and recent work has further characterized the metabolic mechanisms involved. In healthy people eating a traditional diet, including carbohydrates, fats and protein, the major energy substrate in brain is glucose. Cytosolic glucose metabolism generates small amounts of energy, but oxidative glucose metabolism in the mitochondria generates most ATP, in addition to biosynthetic precursors in cells. Energy is crucial for the brain to signal "normally," while loss of energy can contribute to seizure generation by destabilizing membrane potentials and signaling in the chronic epileptic brain. Here we summarize the known biochemical mechanisms that contribute to the disturbance in oxidative glucose metabolism in epilepsy, including decreases in glucose transport, reduced activity of particular steps in the oxidative metabolism of glucose such as pyruvate dehydrogenase activity, and increased anaplerotic need. This knowledge justifies the use of alternative brain fuels as sources of energy, such as ketones, TCA cycle intermediates and precursors as well as even medium chain fatty acids and triheptanoin.
Keywords: anaplerosis; glucose metabolism; medium chain fatty acids; pilocarpine; temporal lobe epilepsy.
Figures
References
-
- Apelt J., Mehlhorn G., Schliebs R. (1999). Insulin-sensitive GLUT4 glucose transporters are colocalized with GLUT3-expressing cells and demonstrate a chemically distinct neuron-specific localization in rat brain. J. Neurosci. Res. 57 693–705. 10.1002/(SICI)1097-4547(19990901)57:5<693::AID-JNR11>3.0.CO;2-X - DOI - PubMed
-
- Bagga P., Behar K. L., Mason G. F., De Feyter H. M., Rothman D. L., Patel A. B. (2014). Characterization of cerebral glutamine uptake from blood in the mouse brain: implications for metabolic modeling of 13C NMR data. J. Cereb Blood Flow Metab. 34 1666–1672. 10.1038/jcbfm.2014.129 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources