

Turn Back the TIMe: Targeting Tumor Infiltrating Myeloid Cells to Revert Cancer Progression
- PMID: 30233579
- PMCID: PMC6127274
- DOI: 10.3389/fimmu.2018.01977
Turn Back the TIMe: Targeting Tumor Infiltrating Myeloid Cells to Revert Cancer Progression
Abstract
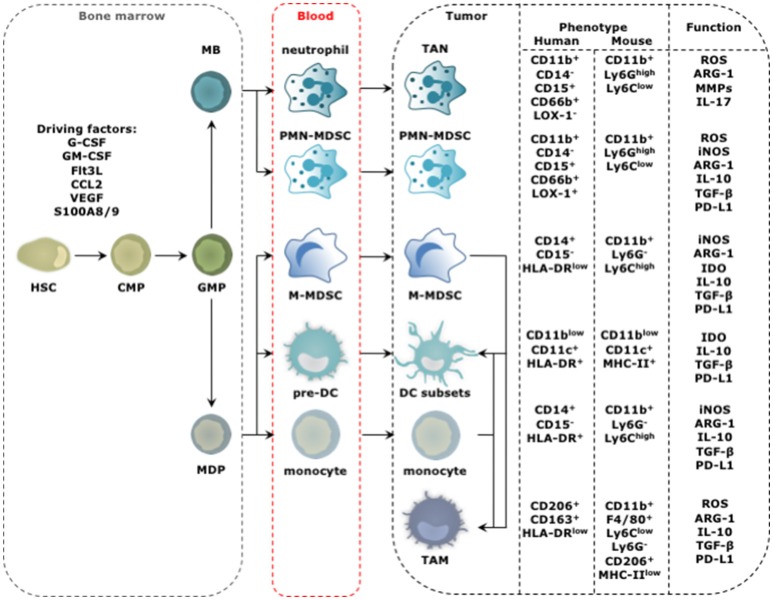
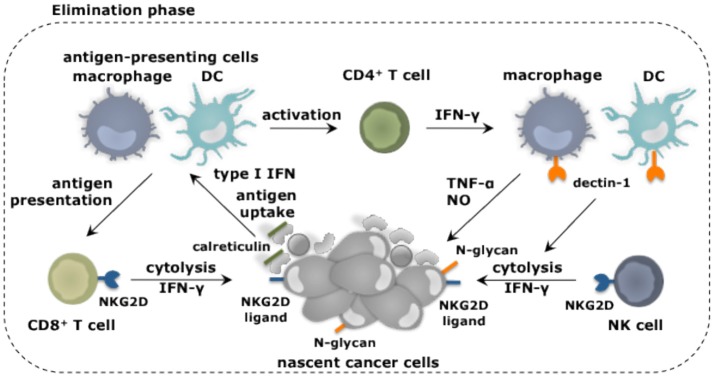
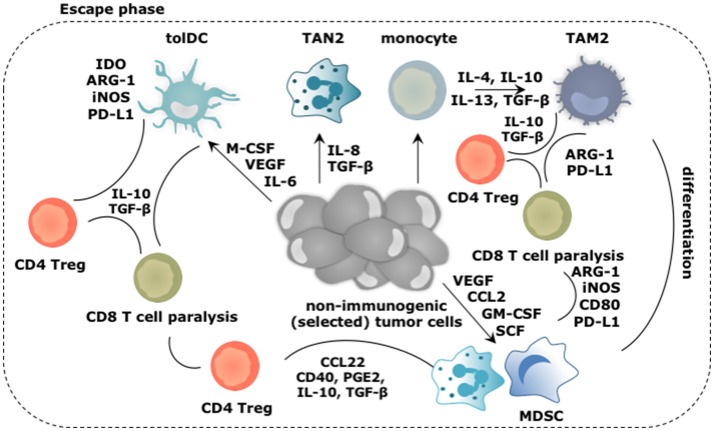
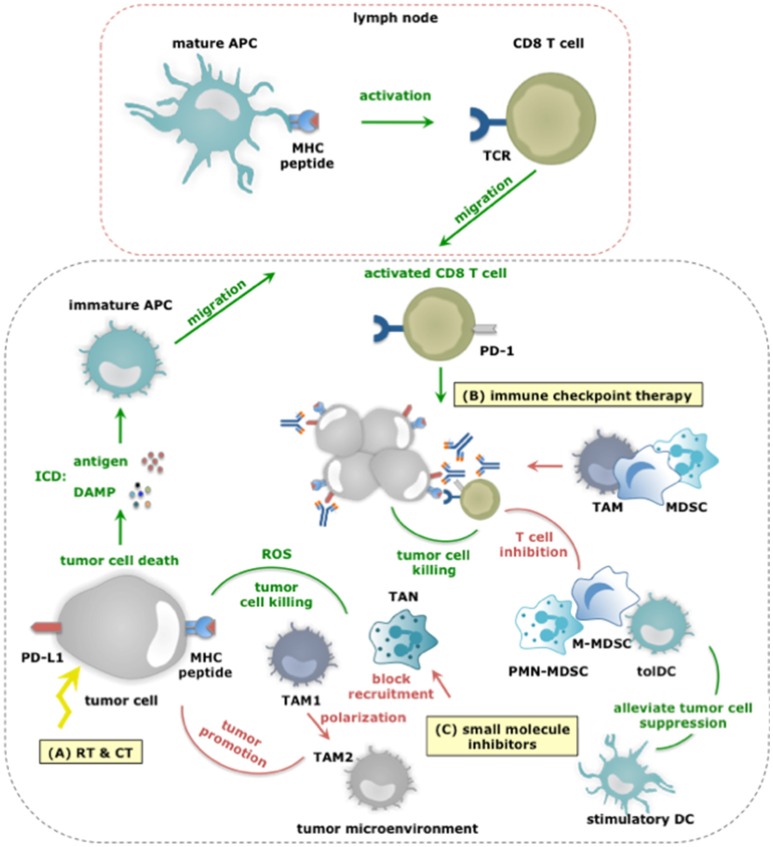
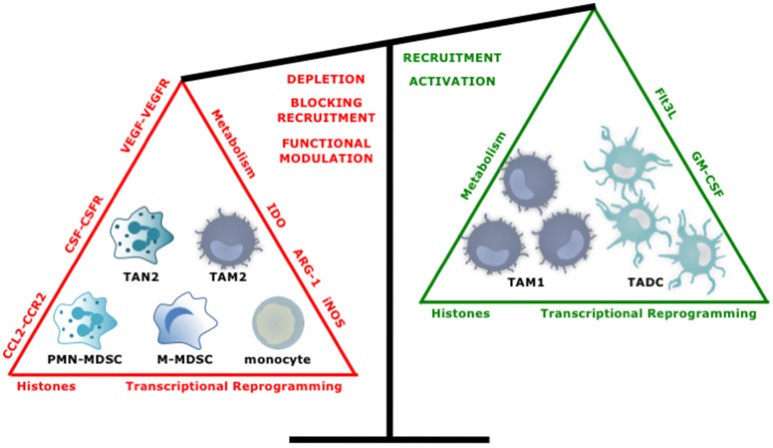
Tumor cells frequently produce soluble factors that favor myelopoiesis and recruitment of myeloid cells to the tumor microenvironment (TME). Consequently, the TME of many cancer types is characterized by high infiltration of monocytes, macrophages, dendritic cells and granulocytes. Experimental and clinical studies show that most myeloid cells are kept in an immature state in the TME. These studies further show that tumor-derived factors mold these myeloid cells into cells that support cancer initiation and progression, amongst others by enabling immune evasion, tumor cell survival, proliferation, migration and metastasis. The key role of myeloid cells in cancer is further evidenced by the fact that they negatively impact on virtually all types of cancer therapy. Therefore, tumor-associated myeloid cells have been designated as the culprits in cancer. We review myeloid cells in the TME with a focus on the mechanisms they exploit to support cancer cells. In addition, we provide an overview of approaches that are under investigation to deplete myeloid cells or redirect their function, as these hold promise to overcome resistance to current cancer therapies.
Keywords: cancer; dendritic cell; immature myeloid cell; macrophage; myeloid-derived suppressor cell; tumor microenvironment.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases