

Molecular Dynamics Simulation for All
- PMID: 30236283
- PMCID: PMC6209097
- DOI: 10.1016/j.neuron.2018.08.011
Molecular Dynamics Simulation for All
Abstract
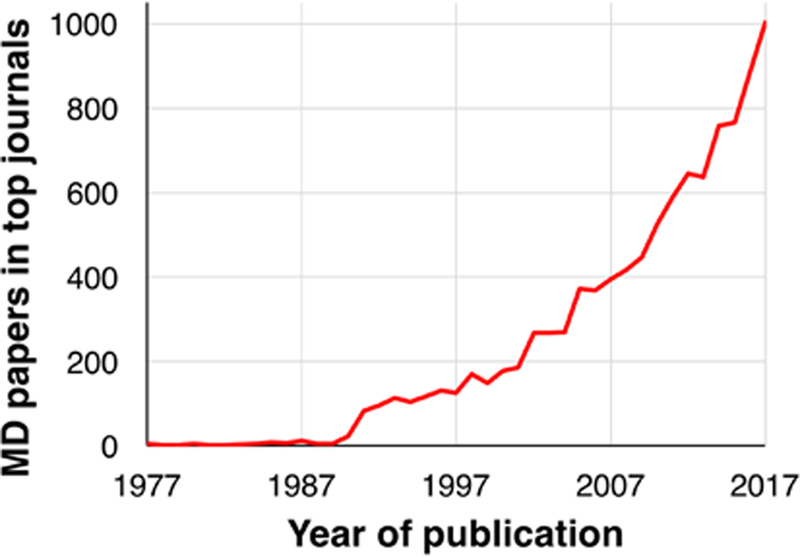
The impact of molecular dynamics (MD) simulations in molecular biology and drug discovery has expanded dramatically in recent years. These simulations capture the behavior of proteins and other biomolecules in full atomic detail and at very fine temporal resolution. Major improvements in simulation speed, accuracy, and accessibility, together with the proliferation of experimental structural data, have increased the appeal of biomolecular simulation to experimentalists-a trend particularly noticeable in, although certainly not limited to, neuroscience. Simulations have proven valuable in deciphering functional mechanisms of proteins and other biomolecules, in uncovering the structural basis for disease, and in the design and optimization of small molecules, peptides, and proteins. Here we describe, in practical terms, the types of information MD simulations can provide and the ways in which they typically motivate further experimental work.
Keywords: MD simulations; allostery; biomolecular simulation; conformational change; drug design; drug discovery; experimental design; protein; structural biology.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of Interests
The authors declare no competing interests.
Figures
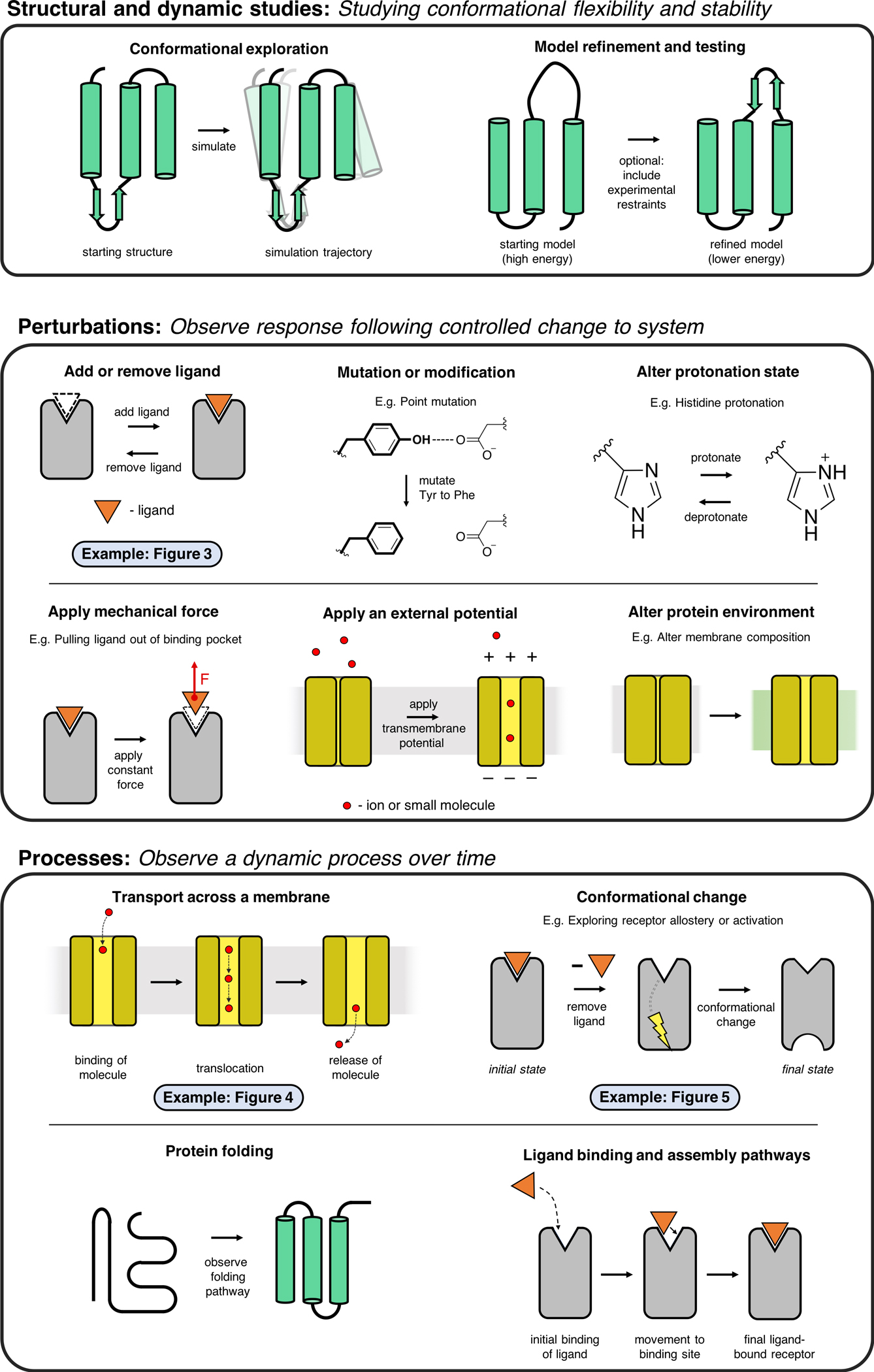
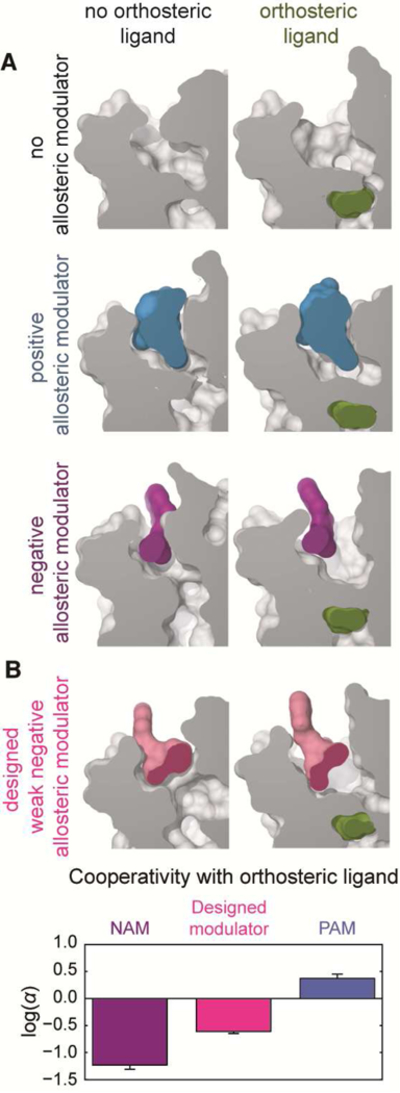
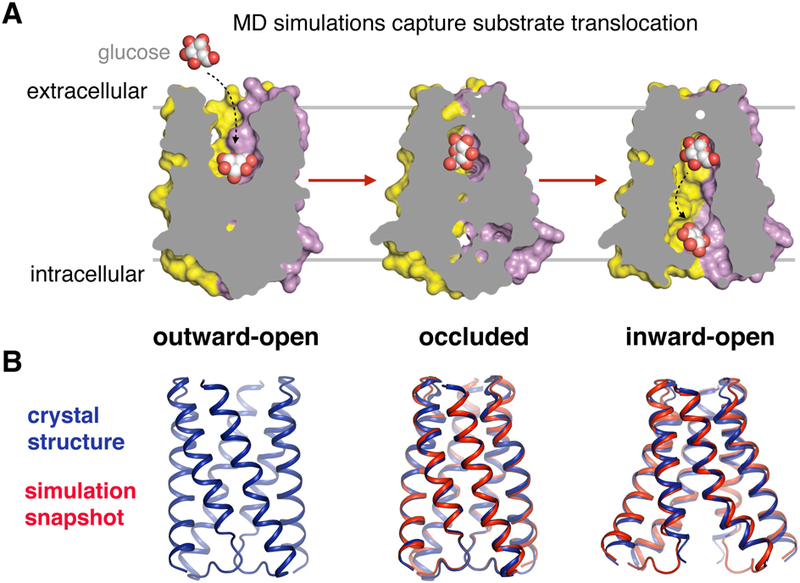
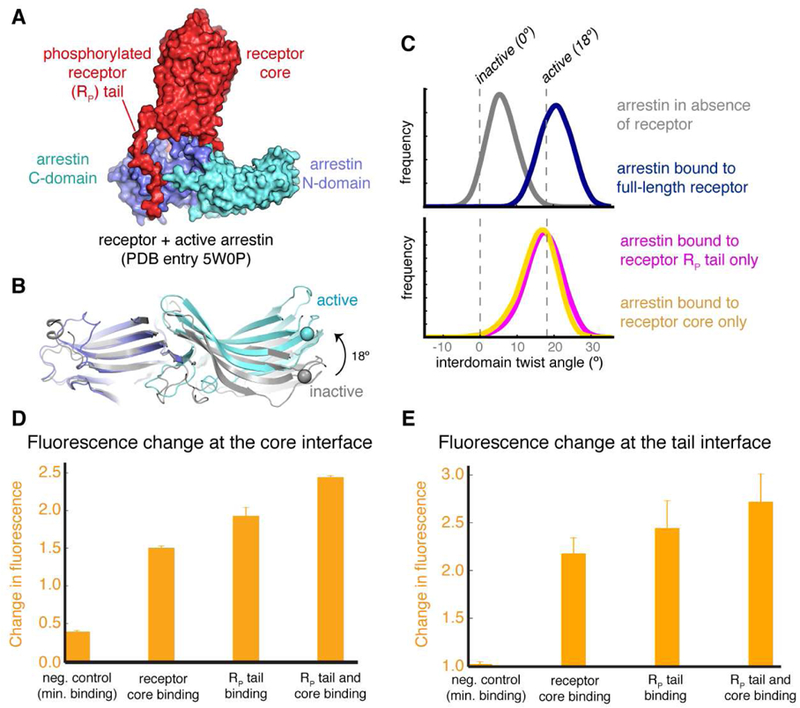
References
-
- Abraham MJ, Murtola T, Schulz R, Páll S, Smith JC, Hess B, and Lindahl E (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1, 19–25.
-
- Alder BJ, and Wainwright TE (1957). Phase Transition for a Hard Sphere System. J Chem Phys 27, 1208–1209.
-
- Arkin IT, Xu H, Jensen MØ, Arbely E, Bennett ER, Bowers KJ, Chow E, Dror RO, Eastwood MP, Flitman-Tene R, et al. (2007). Mechanism of Na+/H+ antiporting. Science 317, 799–803. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
