

Helicobacter pylori infection and inflammatory bowel disease: a crosstalk between upper and lower digestive tract
- PMID: 30237392
- PMCID: PMC6148320
- DOI: 10.1038/s41419-018-0982-2
Helicobacter pylori infection and inflammatory bowel disease: a crosstalk between upper and lower digestive tract
Abstract
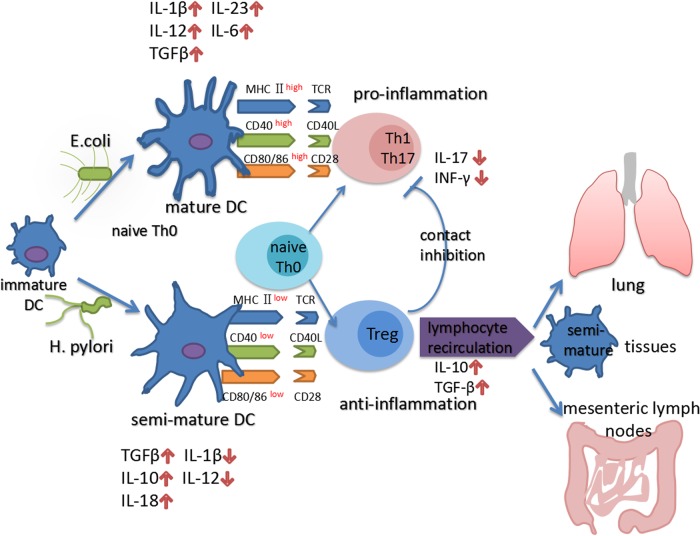
Helicobacter pylori has coexisted with humans for approximately 60,000 years and greater than 50% of the global population is infected with H. pylori. H. pylori was successfully cultured in vitro in 1983 and studies of H. pylori have achieved substantial advances over the last 35 years. Since then, H. pylori has been characterized as the primary pathogenic factor for chronic gastritis, peptic ulcer, and gastric malignancy. Numerous patients have received H. pylori eradication treatment, but only 1-2% of H. pylori-infected individuals ultimately develop gastric cancer. Recently, numerous epidemiological and basic experimental studies suggested a role for chronic H. pylori infection in protecting against inflammatory bowel disease (IBD) by inducing systematic immune tolerance and suppressing inflammatory responses. Here we summarize the current research progress on the association between H. pylori and IBD, and further describe the detailed molecular mechanism underlying H. pylori-induced dendritic cells (DCs) with the tolerogenic phenotype and immunosuppressive regulatory T cells (Tregs). Based on the potential protective role of H. pylori infection on IBD, we suggest that the interaction between H. pylori and the host is complicated, and H. pylori eradication treatment should be administered with caution, especially for children and young adults.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
