

Pulmonary Metagenomic Sequencing Suggests Missed Infections in Immunocompromised Children
- PMID: 30239621
- PMCID: PMC6784263
- DOI: 10.1093/cid/ciy802
Pulmonary Metagenomic Sequencing Suggests Missed Infections in Immunocompromised Children
Abstract
Background: Despite improved diagnostics, pulmonary pathogens in immunocompromised children frequently evade detection, leading to significant mortality. Therefore, we aimed to develop a highly sensitive metagenomic next-generation sequencing (mNGS) assay capable of evaluating the pulmonary microbiome and identifying diverse pathogens in the lungs of immunocompromised children.
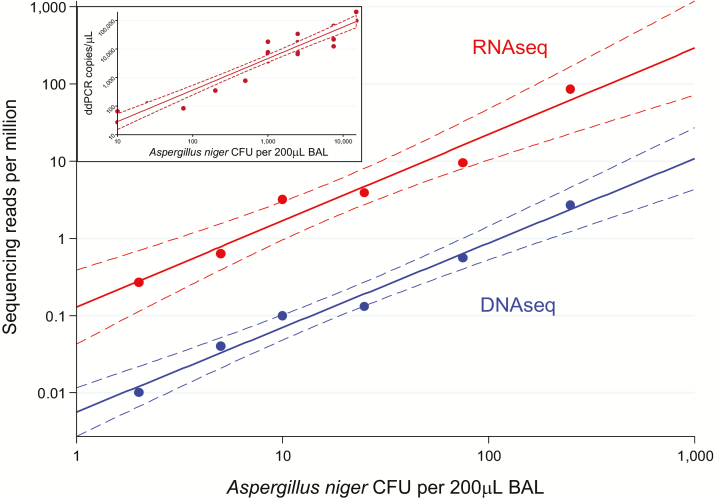
Methods: We collected 41 lower respiratory specimens from 34 immunocompromised children undergoing evaluation for pulmonary disease at 3 children's hospitals from 2014-2016. Samples underwent mechanical homogenization, parallel RNA/DNA extraction, and metagenomic sequencing. Sequencing reads were aligned to the National Center for Biotechnology Information nucleotide reference database to determine taxonomic identities. Statistical outliers were determined based on abundance within each sample and relative to other samples in the cohort.
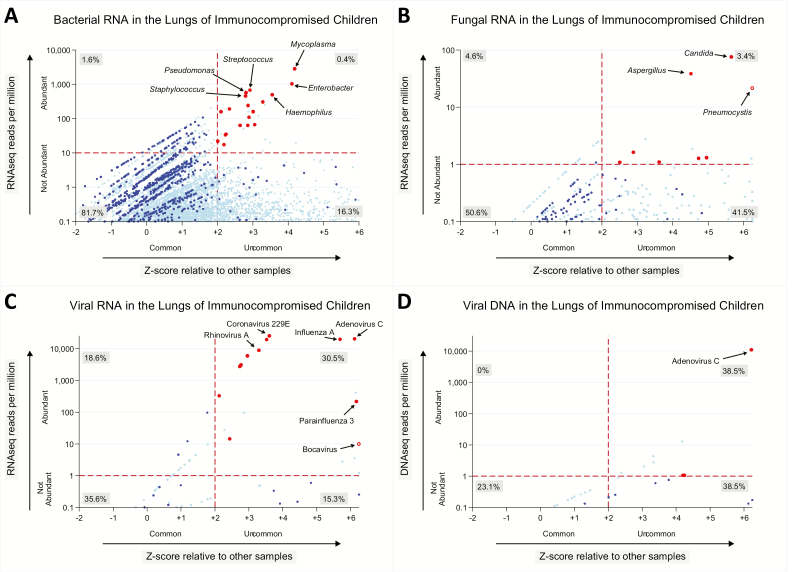
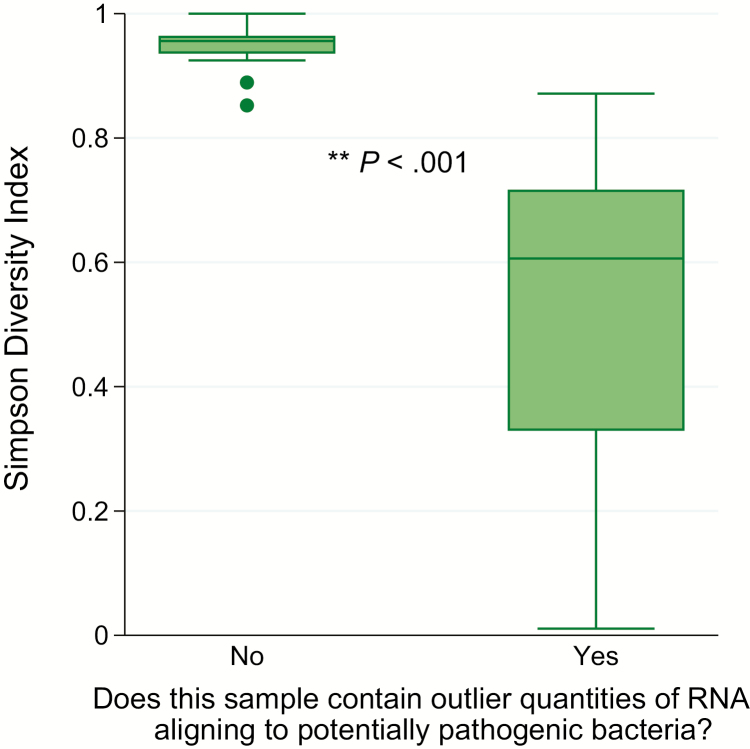
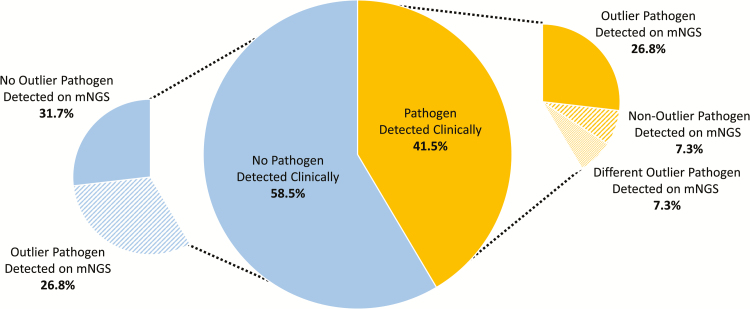
Results: We identified a rich cross-domain pulmonary microbiome that contained bacteria, fungi, RNA viruses, and DNA viruses in each patient. Potentially pathogenic bacteria were ubiquitous among samples but could be distinguished as possible causes of disease by parsing for outlier organisms. Samples with bacterial outliers had significantly depressed alpha-diversity (median, 0.61; interquartile range [IQR], 0.33-0.72 vs median, 0.96; IQR, 0.94-0.96; P < .001). Potential pathogens were detected in half of samples previously negative by clinical diagnostics, demonstrating increased sensitivity for missed pulmonary pathogens (P < .001).
Conclusions: An optimized mNGS assay for pulmonary microbes demonstrates significant inoculation of the lower airways of immunocompromised children with diverse bacteria, fungi, and viruses. Potential pathogens can be identified based on absolute and relative abundance. Ongoing investigation is needed to determine the pathogenic significance of outlier microbes in the lungs of immunocompromised children with pulmonary disease.
Keywords: immunocompromised host; intensive care units; metagenomics; microbiota; pediatric; respiratory tract infections.
© The Author(s) 2018. Published by Oxford University Press for the Infectious Diseases Society of America. All rights reserved. For permissions, e-mail: journals.permissions@oup.com.
Figures
Similar articles
-
Detection of Pulmonary Infectious Pathogens From Lung Biopsy Tissues by Metagenomic Next-Generation Sequencing.Front Cell Infect Microbiol. 2018 Jun 25;8:205. doi: 10.3389/fcimb.2018.00205. eCollection 2018. Front Cell Infect Microbiol. 2018. PMID: 29988504 Free PMC article.
-
Improving Pulmonary Infection Diagnosis with Metagenomic Next Generation Sequencing.Front Cell Infect Microbiol. 2021 Jan 26;10:567615. doi: 10.3389/fcimb.2020.567615. eCollection 2020. Front Cell Infect Microbiol. 2021. PMID: 33585263 Free PMC article.
-
Retrospective Validation of a Metagenomic Sequencing Protocol for Combined Detection of RNA and DNA Viruses Using Respiratory Samples from Pediatric Patients.J Mol Diagn. 2020 Feb;22(2):196-207. doi: 10.1016/j.jmoldx.2019.10.007. Epub 2019 Dec 16. J Mol Diagn. 2020. PMID: 31837435 Free PMC article.
-
Metagenomics next-generation sequencing tests take the stage in the diagnosis of lower respiratory tract infections.J Adv Res. 2021 Sep 29;38:201-212. doi: 10.1016/j.jare.2021.09.012. eCollection 2022 May. J Adv Res. 2021. PMID: 35572406 Free PMC article. Review.
-
Airway microbial metagenomics.Microbes Infect. 2018 Oct-Nov;20(9-10):536-542. doi: 10.1016/j.micinf.2017.12.002. Epub 2017 Dec 26. Microbes Infect. 2018. PMID: 29287982 Review.
Cited by
-
How We Treat Fever and Hypotension in Pediatric Hematopoietic Cell Transplant Patients.Front Oncol. 2020 Sep 16;10:581447. doi: 10.3389/fonc.2020.581447. eCollection 2020. Front Oncol. 2020. PMID: 33042850 Free PMC article. Review.
-
Metagenomics for neurological infections - expanding our imagination.Nat Rev Neurol. 2020 Oct;16(10):547-556. doi: 10.1038/s41582-020-0374-y. Epub 2020 Jul 13. Nat Rev Neurol. 2020. PMID: 32661342 Free PMC article. Review.
-
BALF metagenomic next-generation sequencing analysis in hematological malignancy patients with suspected pulmonary infection: clinical significance of negative results.Front Med (Lausanne). 2023 Jun 28;10:1195629. doi: 10.3389/fmed.2023.1195629. eCollection 2023. Front Med (Lausanne). 2023. PMID: 37457591 Free PMC article.
-
Extracorporeal membrane oxygenation in children receiving haematopoietic cell transplantation and immune effector cell therapy: an international and multidisciplinary consensus statement.Lancet Child Adolesc Health. 2022 Feb;6(2):116-128. doi: 10.1016/S2352-4642(21)00336-9. Epub 2021 Dec 9. Lancet Child Adolesc Health. 2022. PMID: 34895512 Free PMC article. Review.
-
Utilizing metagenomic next-generation sequencing for diagnosis and lung microbiome probing of pediatric pneumonia through bronchoalveolar lavage fluid in pediatric intensive care unit: results from a large real-world cohort.Front Cell Infect Microbiol. 2023 Aug 15;13:1200806. doi: 10.3389/fcimb.2023.1200806. eCollection 2023. Front Cell Infect Microbiol. 2023. PMID: 37655299 Free PMC article.
References
-
- Center for International Blood and Marrow Transplant, a contractor for the C.W. Bill Young Cell Transplantation Program operated through the U. S. Department of Health and Human Services, Health Resources and Services Administration, Healthcare Systems Bureau. U.S. transplant data by age, number of transplants reported in 2016. Available at: https://bloodcell.transplant.hrsa.gov/research/transplant_data/transplan.... Accessed 1 July 2017.
-
- OPTN/SRTR 2015 annual data report: introduction. Am J Transplant 2017; 17(Suppl 1):11–20. - PubMed
-
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017; 67:7–30. - PubMed
-
- Kaya Z, Weiner DJ, Yilmaz D, Rowan J, Goyal RK. Lung function, pulmonary complications, and mortality after allogeneic blood and marrow transplantation in children. Biol Blood Marrow Transplant 2009; 15:817–26. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
