

Injury, dysbiosis, and filaggrin deficiency drive skin inflammation through keratinocyte IL-1α release
- PMID: 30240702
- PMCID: PMC6424655
- DOI: 10.1016/j.jaci.2018.08.042
Injury, dysbiosis, and filaggrin deficiency drive skin inflammation through keratinocyte IL-1α release
Abstract
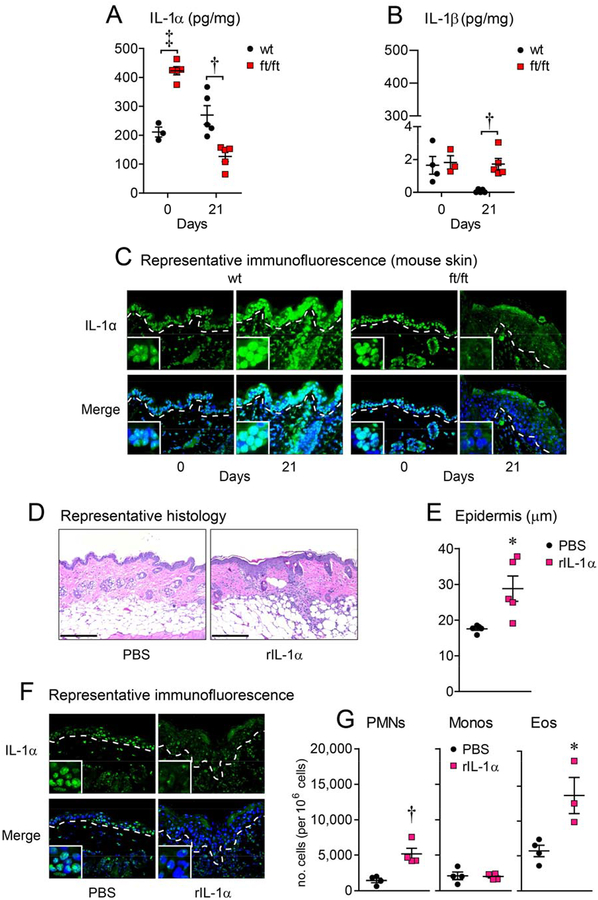
Background: Atopic dermatitis (AD) is associated with epidermal barrier defects, dysbiosis, and skin injury caused by scratching. In particular, the barrier-defective epidermis in patients with AD with loss-of-function filaggrin mutations has increased IL-1α and IL-1β levels, but the mechanisms by which IL-1α, IL-1β, or both are induced and whether they contribute to the aberrant skin inflammation in patients with AD is unknown.
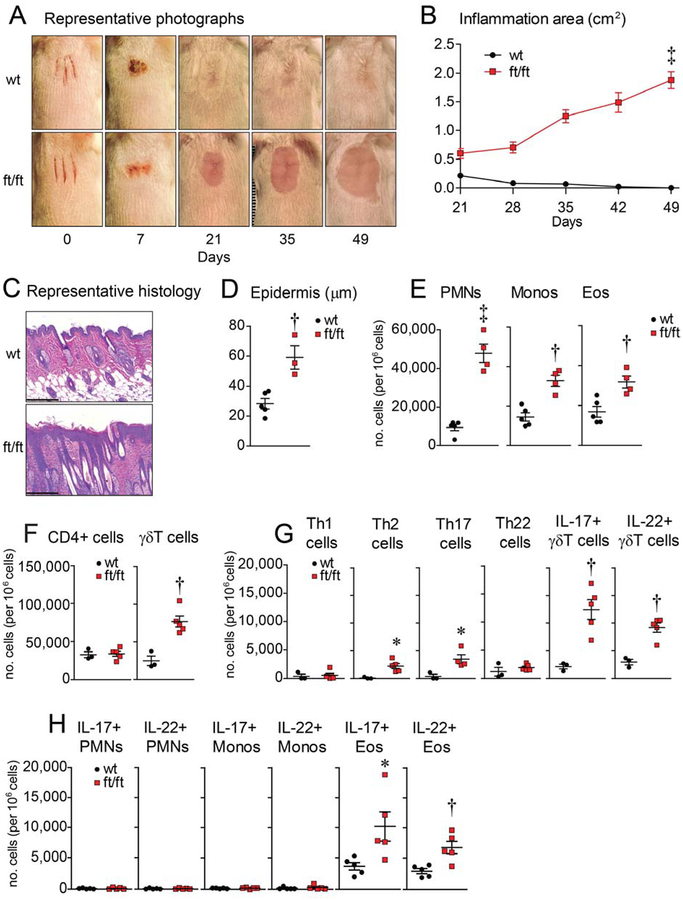
Objective: We sought to determine the mechanisms through which skin injury, dysbiosis, and increased epidermal IL-1α and IL-1β levels contribute to development of skin inflammation in a mouse model of injury-induced skin inflammation in filaggrin-deficient mice without the matted mutation (ft/ft mice).
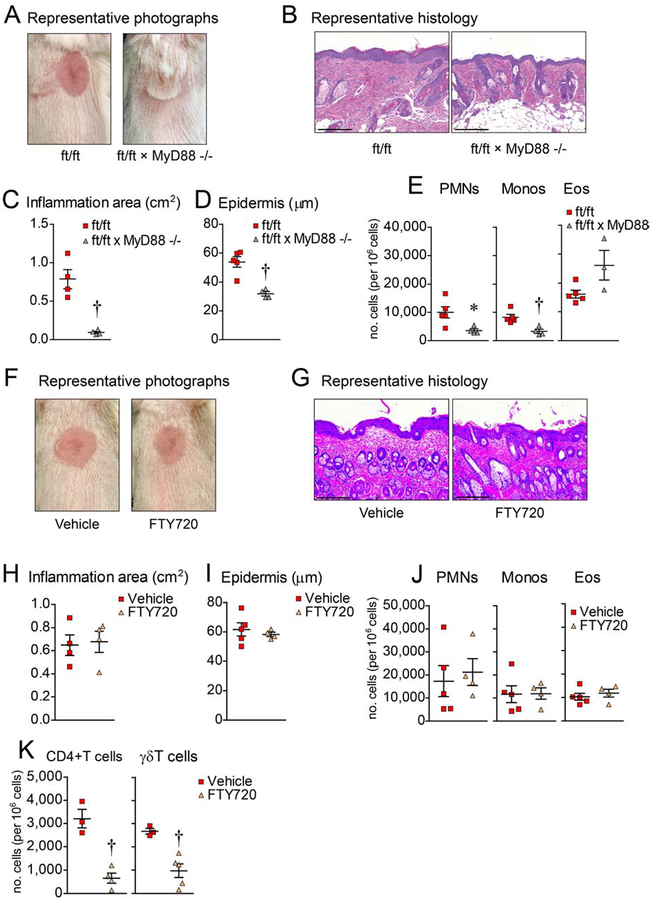
Methods: Skin injury of wild-type, ft/ft, and myeloid differentiation primary response gene-88-deficient ft/ft mice was performed, and ensuing skin inflammation was evaluated by using digital photography, histologic analysis, and flow cytometry. IL-1α and IL-1β protein expression was measured by means of ELISA and visualized by using immunofluorescence and immunoelectron microscopy. Composition of the skin microbiome was determined by using 16S rDNA sequencing.
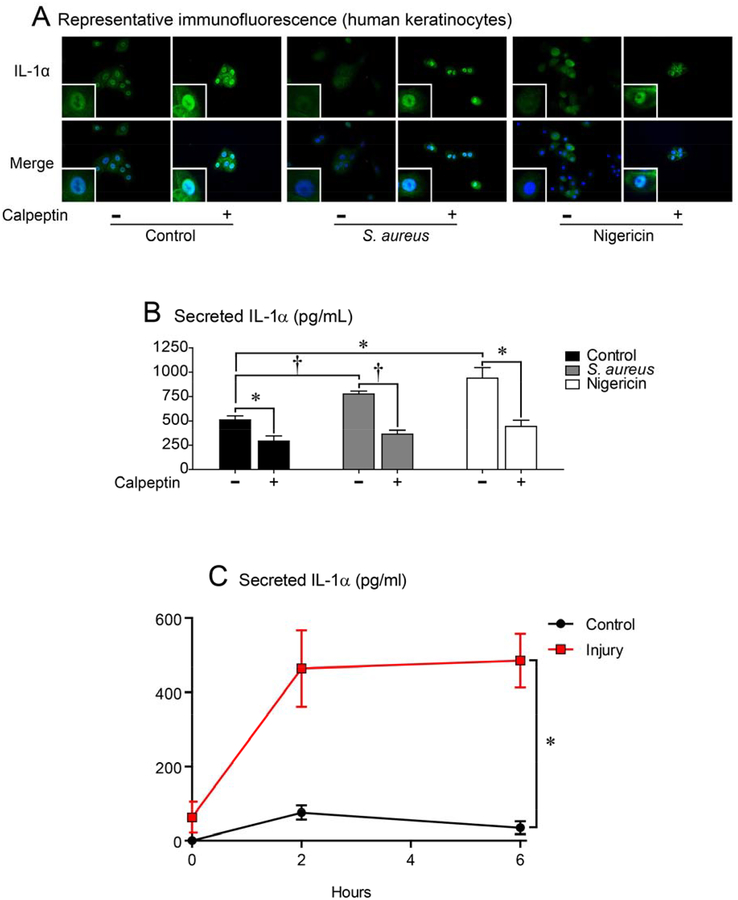
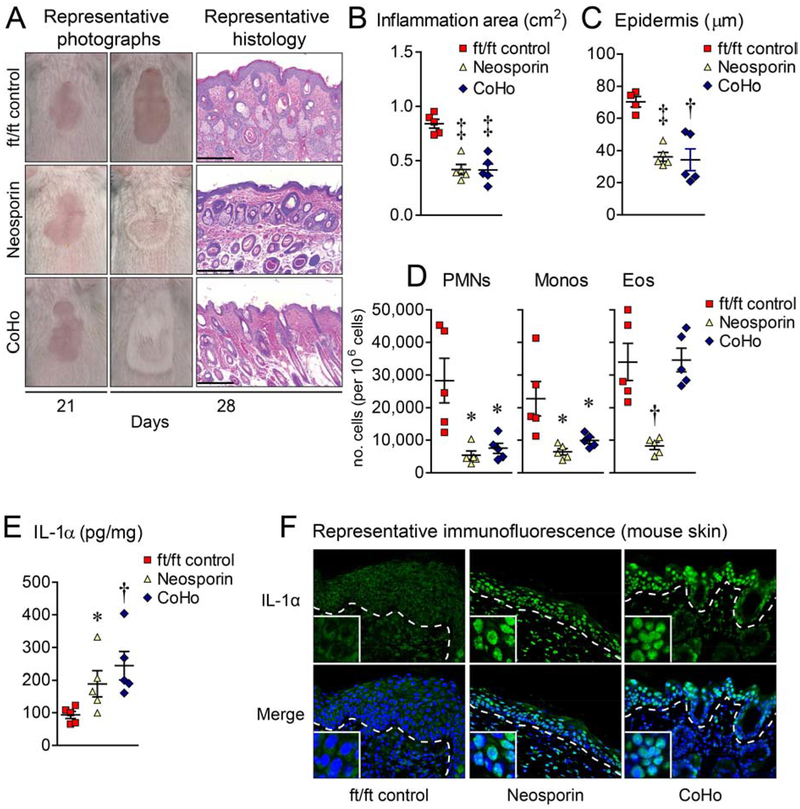
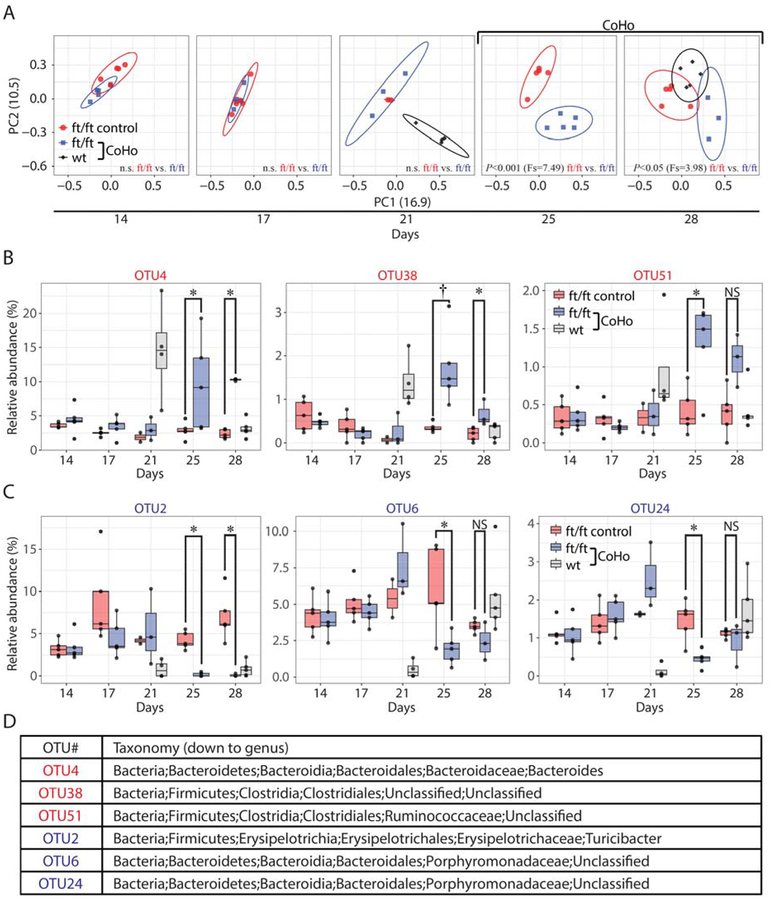
Results: Skin injury of ft/ft mice induced chronic skin inflammation involving dysbiosis-driven intracellular IL-1α release from keratinocytes. IL-1α was necessary and sufficient for skin inflammation in vivo and secreted from keratinocytes by various stimuli in vitro. Topical antibiotics or cohousing of ft/ft mice with unaffected wild-type mice to alter or intermix skin microbiota, respectively, resolved the skin inflammation and restored keratinocyte intracellular IL-1α localization.
Conclusions: Taken together, skin injury, dysbiosis, and filaggrin deficiency triggered keratinocyte intracellular IL-1α release that was sufficient to drive chronic skin inflammation, which has implications for AD pathogenesis and potential therapeutic targets.
Keywords: IL-1α; Skin; atopic dermatitis; filaggrin; inflammation; keratinocytes.
Copyright © 2018 American Academy of Allergy, Asthma & Immunology. All rights reserved.
Conflict of interest statement
Figures


References
-
- Heath WR, Carbone FR. The skin-resident and migratory immune system in steady state and memory: innate lymphocytes, dendritic cells and T cells. Nat Immunol 2013; 14:978–85. - PubMed
-
- Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol 2014; 14:289–301. - PubMed
-
- Drucker AM, Wang AR, Li WQ, Sevetson E, Block JK, Qureshi AA. The Burden of Atopic Dermatitis: Summary of a Report for the National Eczema Association. J Invest Dermatol 2017; 137:26–30. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 AR073665/AR/NIAMS NIH HHS/United States
- UL1 TR000170/TR/NCATS NIH HHS/United States
- K01 AR073924/AR/NIAMS NIH HHS/United States
- R01 AR074846/AR/NIAMS NIH HHS/United States
- R01 AR069502/AR/NIAMS NIH HHS/United States
- R01 AR064297/AR/NIAMS NIH HHS/United States
- R21 AI126896/AI/NIAID NIH HHS/United States
- ZIA HG000180/ImNIH/Intramural NIH HHS/United States
- U19 AI117673/AI/NIAID NIH HHS/United States
- ZIA BC011558/ImNIH/Intramural NIH HHS/United States
- U01 AI147462/AI/NIAID NIH HHS/United States
- R01 AR068280/AR/NIAMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
