Microbiome 101: Studying, Analyzing, and Interpreting Gut Microbiome Data for Clinicians
- PMID: 30240894
- PMCID: PMC6391518
- DOI: 10.1016/j.cgh.2018.09.017
Microbiome 101: Studying, Analyzing, and Interpreting Gut Microbiome Data for Clinicians
Abstract
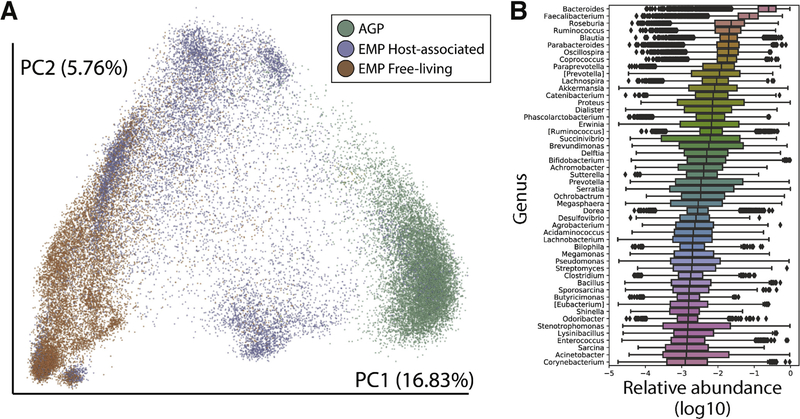
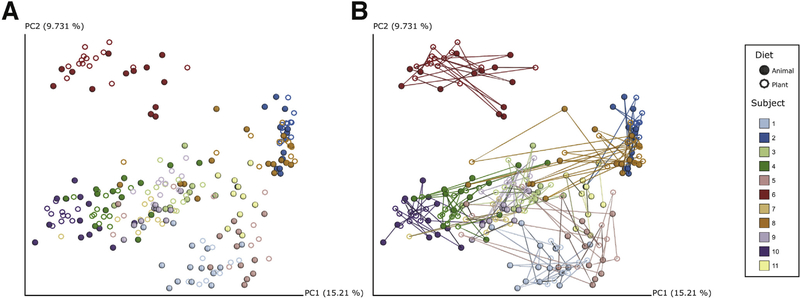
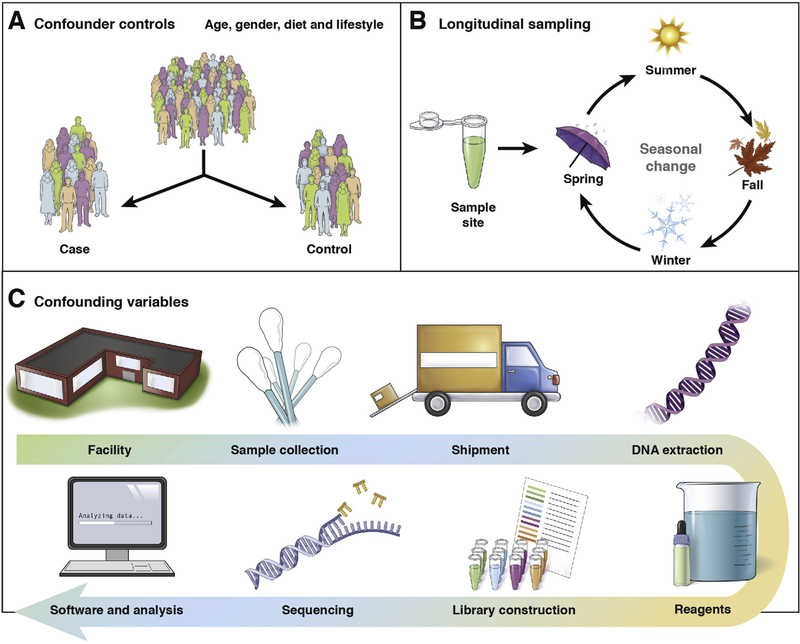
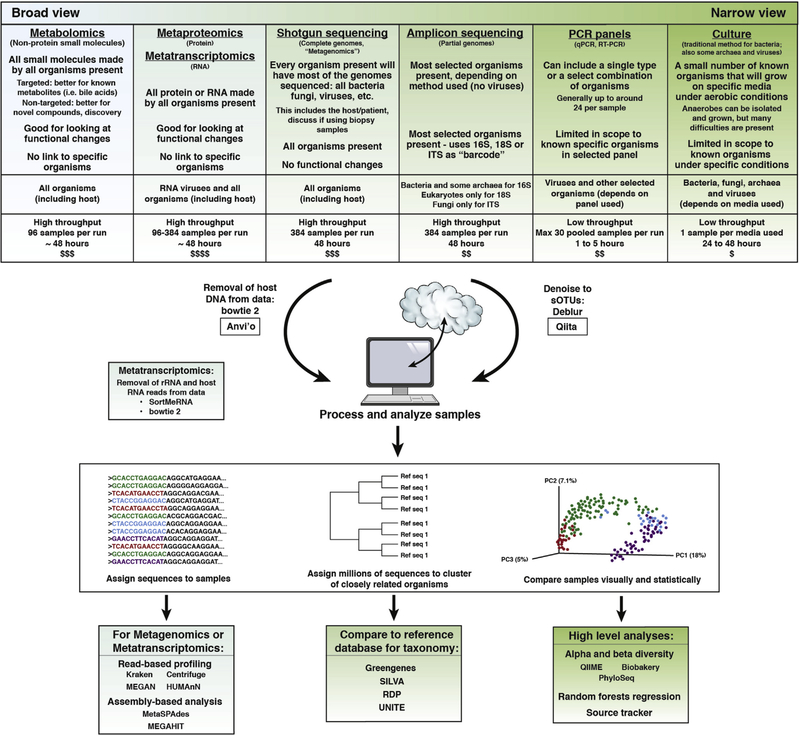
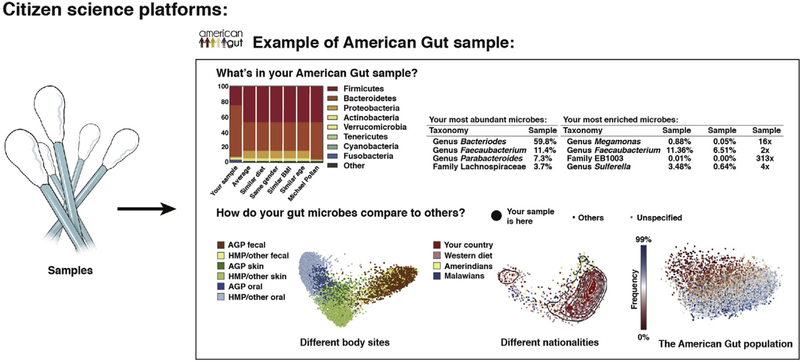
Advances in technical capabilities for reading complex human microbiomes are leading to an explosion of microbiome research, leading in turn to intense interest among clinicians in applying these techniques to their patients. In this review, we discuss the content of the human microbiome, including intersubject and intrasubject variability, considerations of study design including important confounding factors, and different methods in the laboratory and on the computer to read the microbiome and its resulting gene products and metabolites. We highlight several common pitfalls for clinicians, including the expectation that an individual's microbiome will be stable, that diet can induce rapid changes that are large compared with the differences among subjects, that everyone has essentially the same core stool microbiome, and that different laboratory and computational methods will yield essentially the same results. We also highlight the current limitations and future promise of these techniques, with the expectation that an understanding of these considerations will help accelerate the path toward routine clinical application of these techniques developed in research settings.
Keywords: Clinician; Diagnosis; Gut Microbiome; Prognosis; Study Design.
Copyright © 2019 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures