

The genetic architecture of aniridia and Gillespie syndrome
- PMID: 30242502
- PMCID: PMC6710220
- DOI: 10.1007/s00439-018-1934-8
The genetic architecture of aniridia and Gillespie syndrome
Abstract
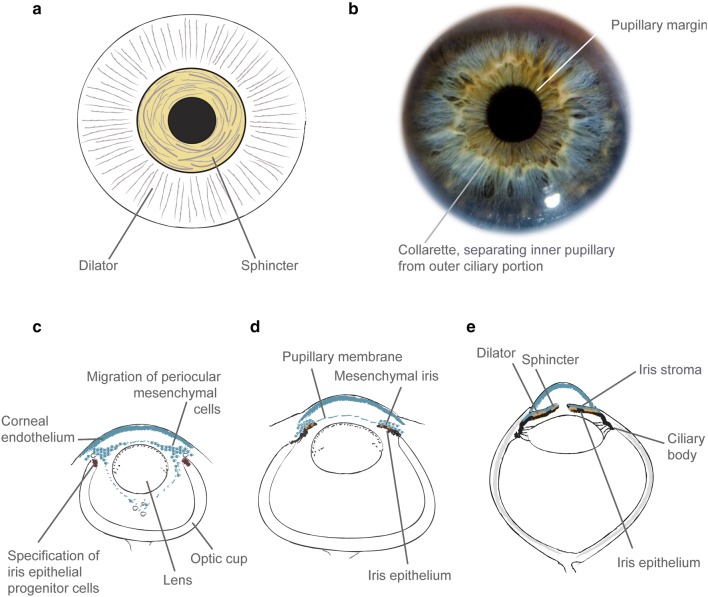
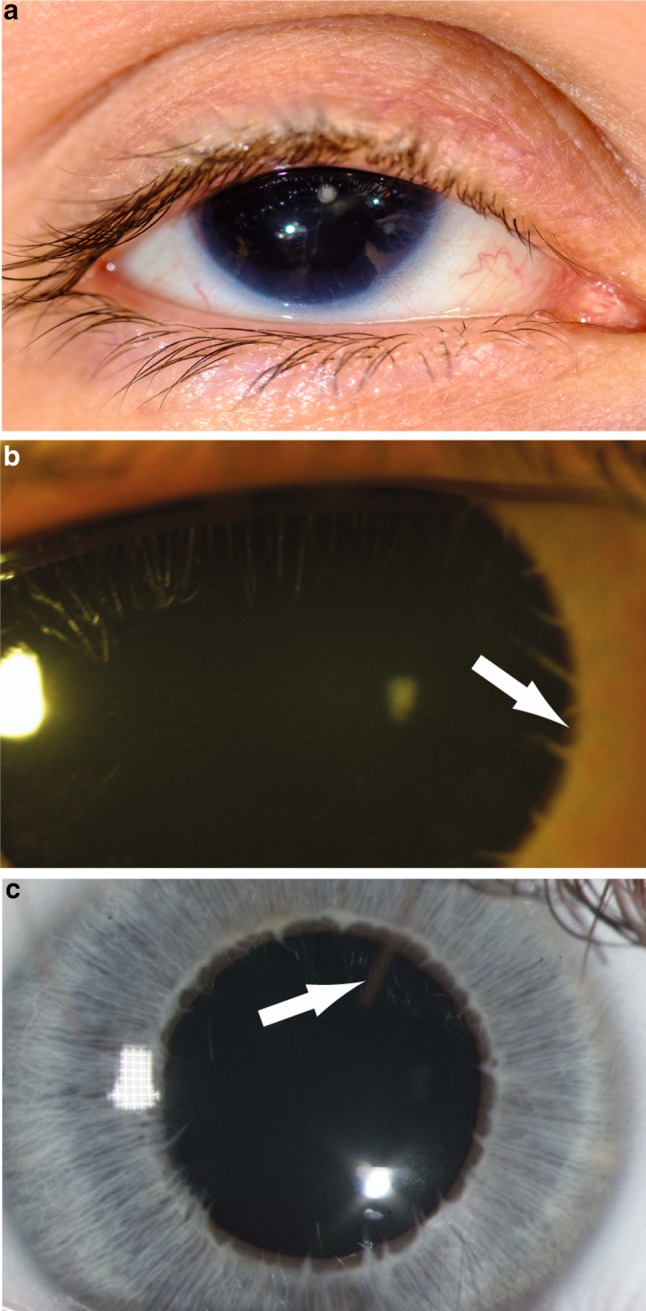
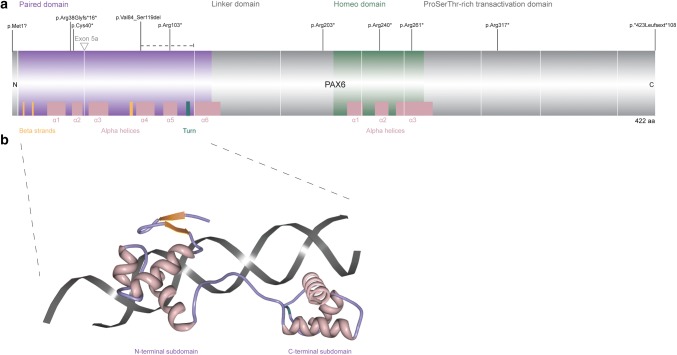
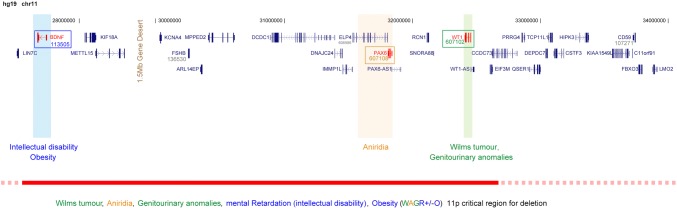
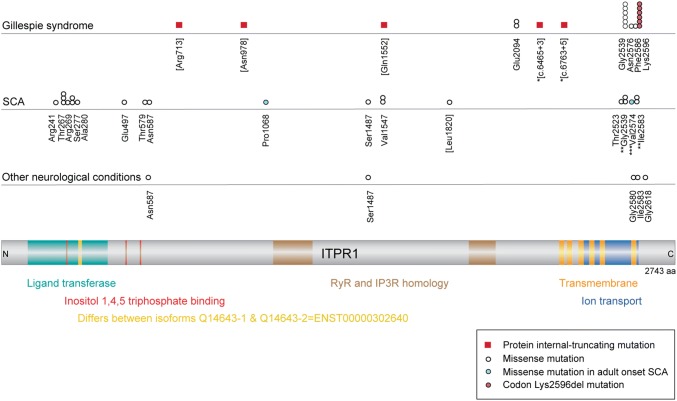
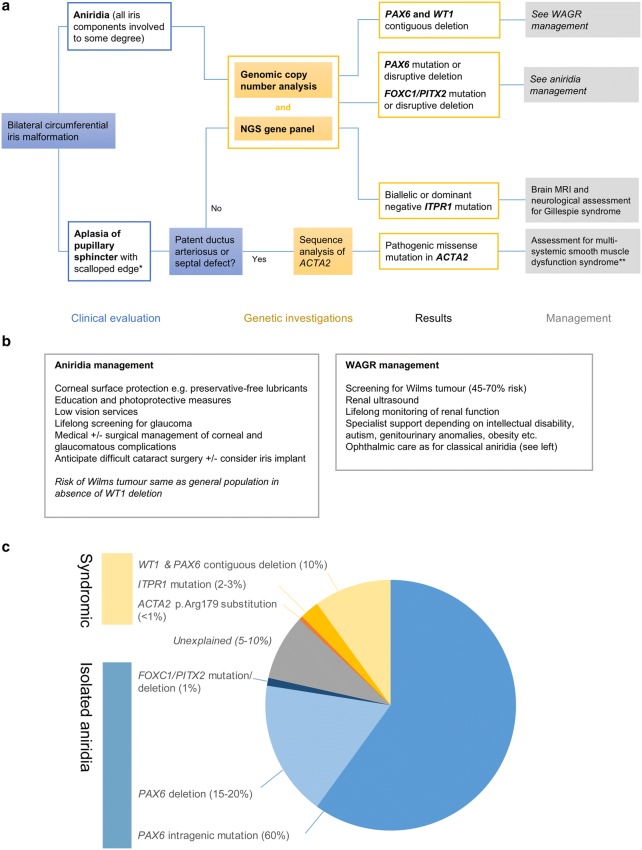
Absence of part or all of the iris, aniridia, is a feature of several genetically distinct conditions. This review focuses on iris development and then the clinical features and molecular genetics of these iris malformations. Classical aniridia, a panocular eye malformation including foveal hypoplasia, is the archetypal phenotype associated with heterozygous PAX6 loss-of-function mutations. Since this was identified in 1991, many genetic mechanisms of PAX6 inactivation have been elucidated, the commonest alleles being intragenic mutations causing premature stop codons, followed by those causing C-terminal extensions. Rarely, aniridia cases are associated with FOXC1, PITX2 and/or their regulatory regions. Aniridia can also occur as a component of many severe global eye malformations. Gillespie syndrome-a triad of partial aniridia, non-progressive cerebellar ataxia and intellectual disability-is phenotypically and genotypically distinct from classical aniridia. The causative gene has recently been identified as ITPR1. The same characteristic Gillespie syndrome-like iris, with aplasia of the pupillary sphincter and a scalloped margin, is seen in ACTA2-related multisystemic smooth muscle dysfunction syndrome. WAGR syndrome (Wilms tumour, aniridia, genitourinary anomalies and mental retardation/intellectual disability), is caused by contiguous deletion of PAX6 and WT1 on chromosome 11p. Deletions encompassing BDNF have been causally implicated in the obesity and intellectual disability associated with the condition. Lastly, we outline a genetic investigation strategy for aniridia in light of recent developments, suggesting an approach based principally on chromosomal array and gene panel testing. This strategy aims to test all known aniridia loci-including the rarer, life-limiting causes-whilst remaining simple and practical.
Conflict of interest statement
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Figures
References
-
- Aalfs CM, Fantes JA, Wenniger-Prick LJJM, et al. Tandem duplication of 11p12–p13 in a child with borderline development delay and eye abnormalities: dose effect of the PAX6 gene product? Am J Med Genet. 1997;73:267–271. - PubMed
-
- Amor DJ. Morbid obesity and hyperphagia in the WAGR syndrome. Clin Dysmorphol. 2002;11:73–74. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
