

Genomic Landscape and Mutational Signatures of Deafness-Associated Genes
- PMID: 30245029
- PMCID: PMC6174355
- DOI: 10.1016/j.ajhg.2018.08.006
Genomic Landscape and Mutational Signatures of Deafness-Associated Genes
Abstract
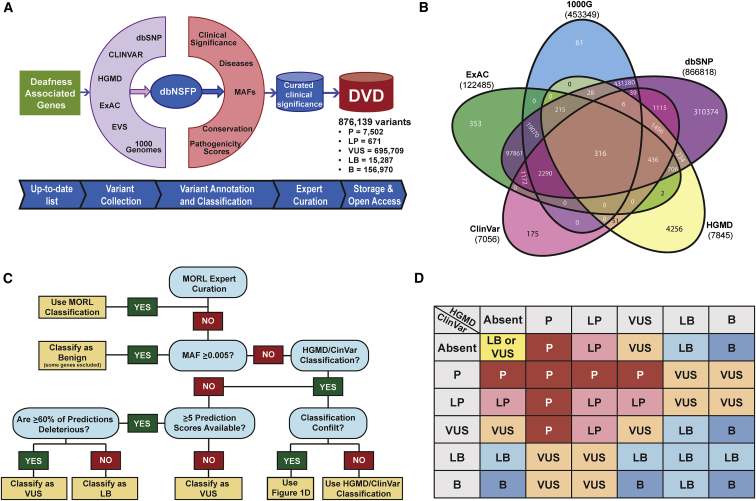
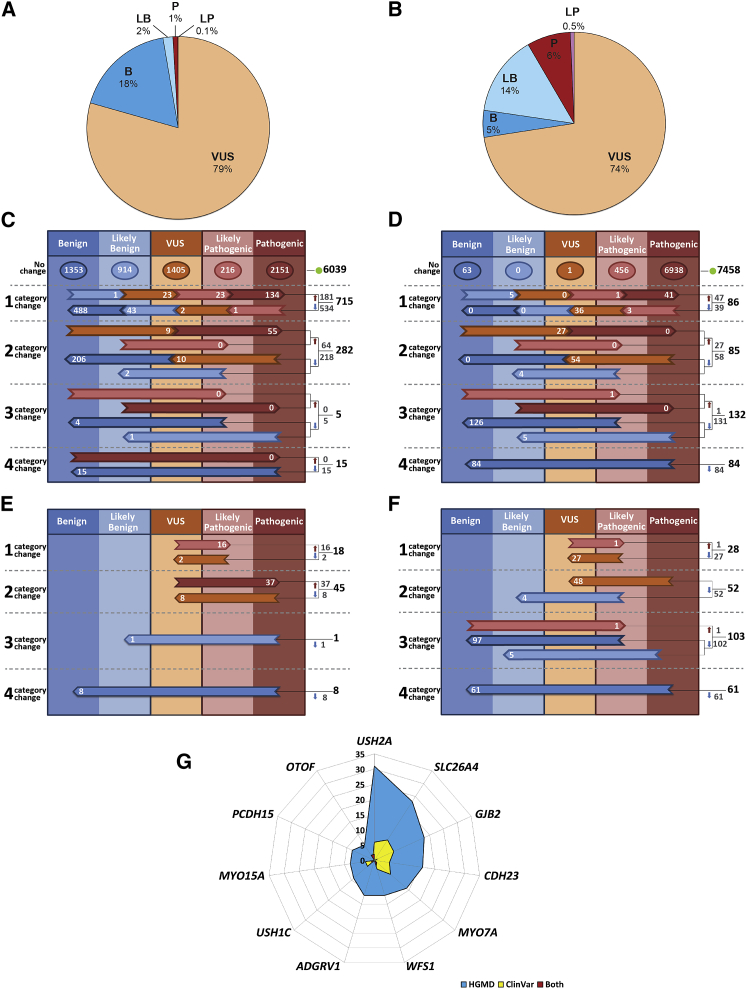
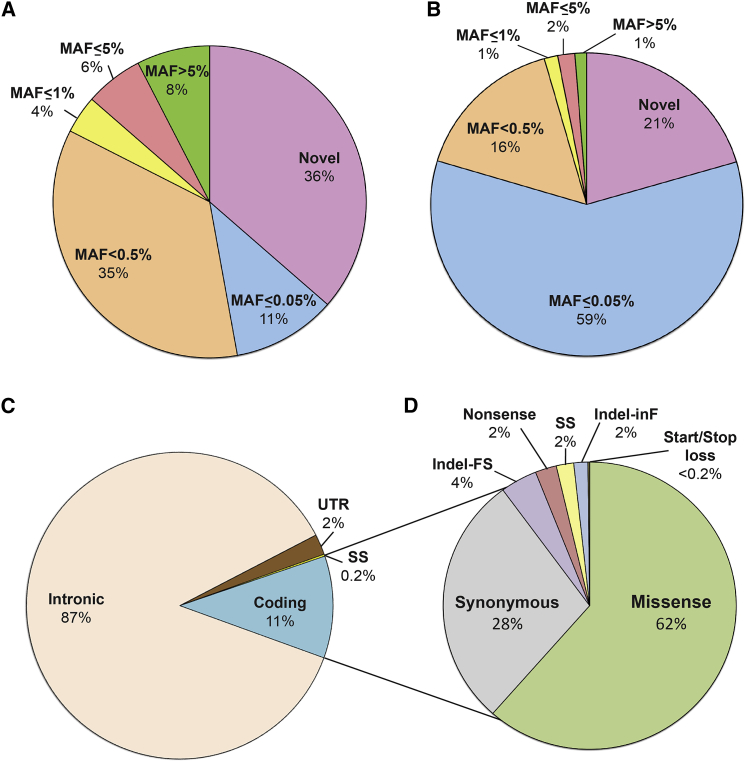
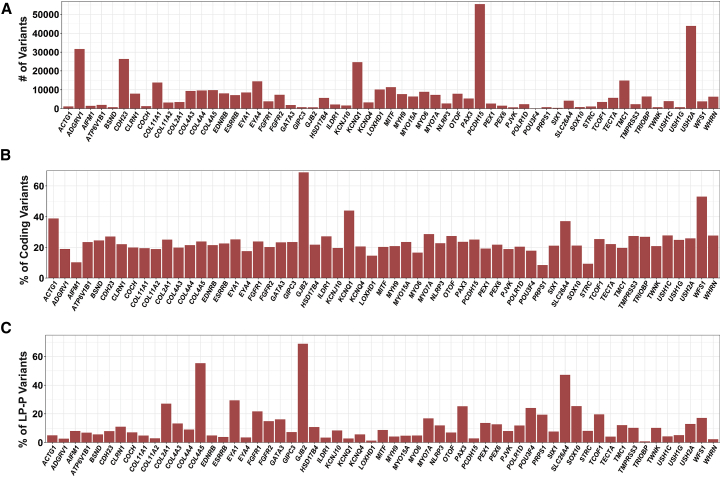
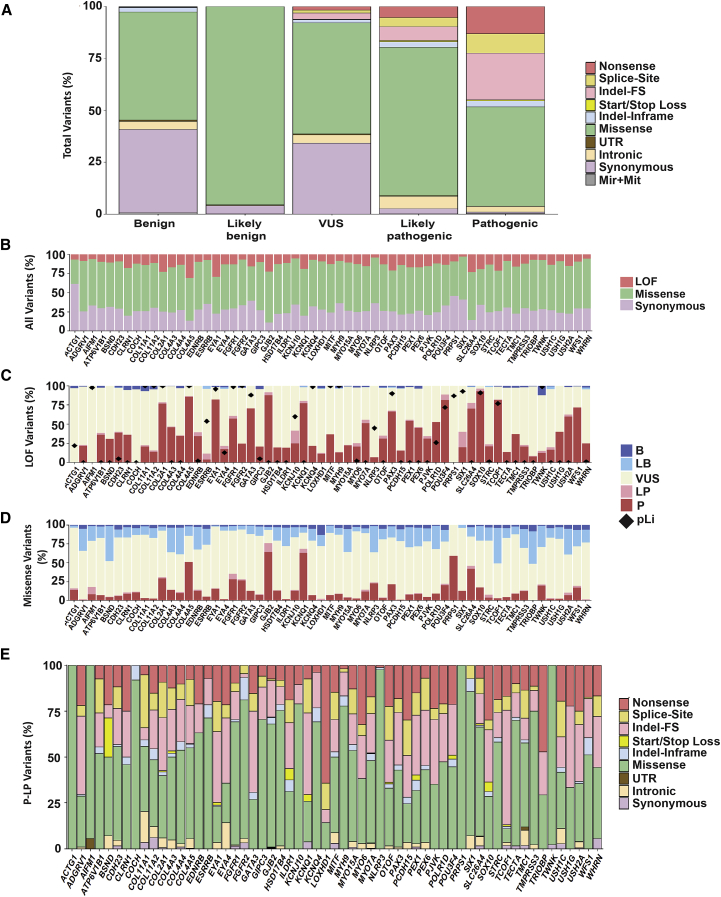
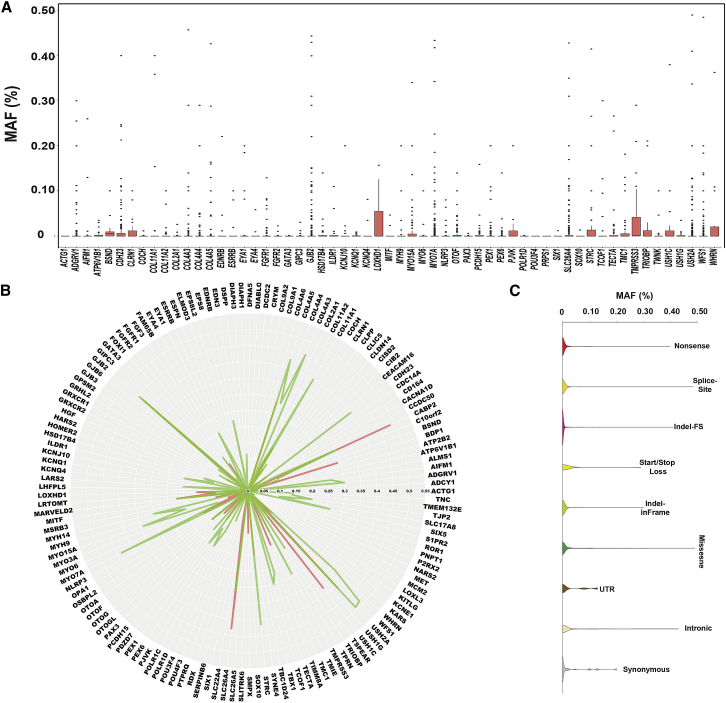
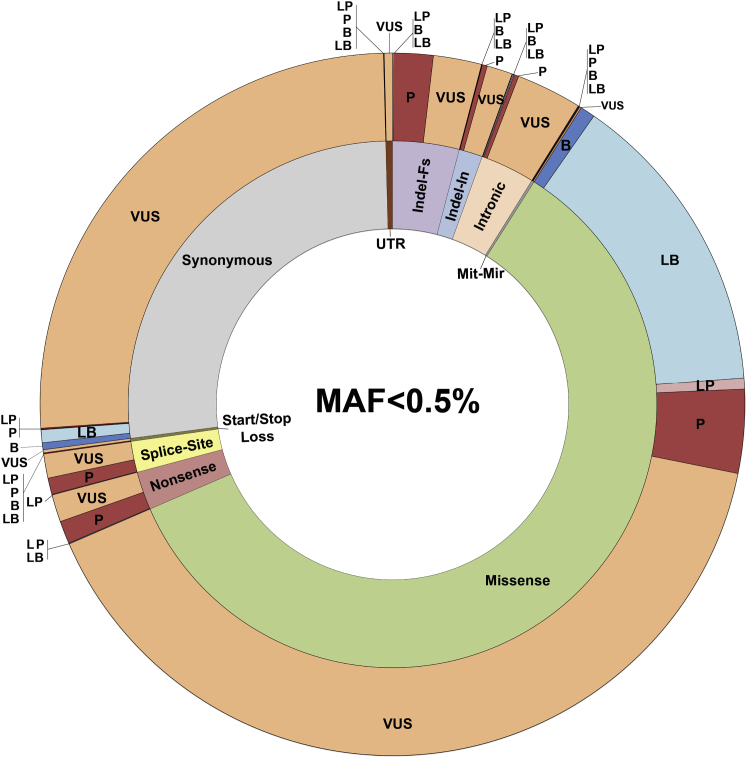
The classification of genetic variants represents a major challenge in the post-genome era by virtue of their extraordinary number and the complexities associated with ascribing a clinical impact, especially for disorders exhibiting exceptional phenotypic, genetic, and allelic heterogeneity. To address this challenge for hearing loss, we have developed the Deafness Variation Database (DVD), a comprehensive, open-access resource that integrates all available genetic, genomic, and clinical data together with expert curation to generate a single classification for each variant in 152 genes implicated in syndromic and non-syndromic deafness. We evaluate 876,139 variants and classify them as pathogenic or likely pathogenic (more than 8,100 variants), benign or likely benign (more than 172,000 variants), or of uncertain significance (more than 695,000 variants); 1,270 variants are re-categorized based on expert curation and in 300 instances, the change is of medical significance and impacts clinical care. We show that more than 96% of coding variants are rare and novel and that pathogenicity is driven by minor allele frequency thresholds, variant effect, and protein domain. The mutational landscape we define shows complex gene-specific variability, making an understanding of these nuances foundational for improved accuracy in variant interpretation in order to enhance clinical decision making and improve our understanding of deafness biology.
Keywords: database; deafness; genetic variant; genomic landscape; mutational signature; precision medicine; variant classification.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W.W., Hegde M., Lyon E., Spector E., ACMG Laboratory Quality Assurance Committee Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–424. - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
