

Splice-altering variant in COL11A1 as a cause of nonsyndromic hearing loss DFNA37
- PMID: 30245514
- PMCID: PMC6431578
- DOI: 10.1038/s41436-018-0285-0
Splice-altering variant in COL11A1 as a cause of nonsyndromic hearing loss DFNA37
Abstract
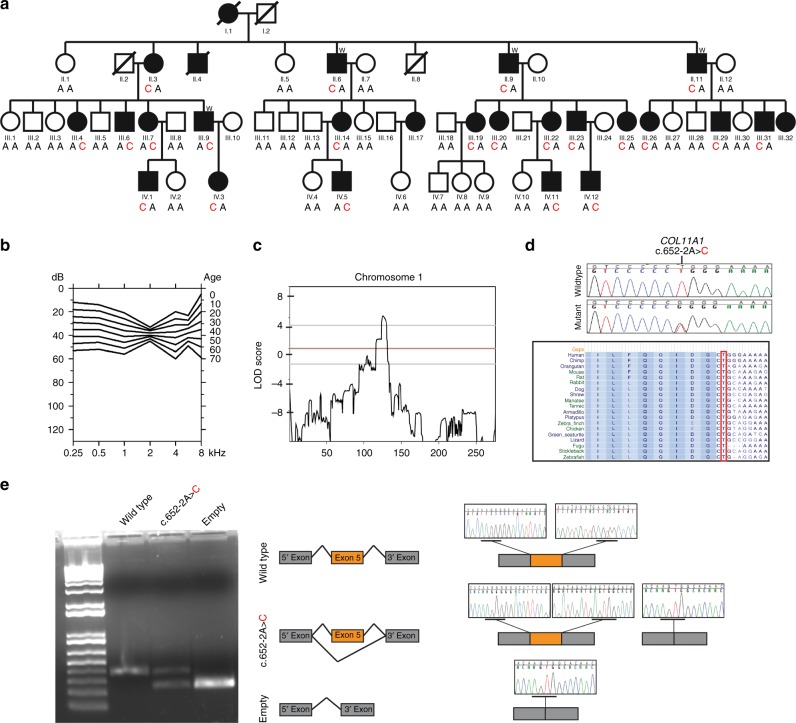
Purpose: The aim of this study was to determine the genetic cause of autosomal dominant nonsyndromic hearing loss segregating in a multigenerational family.
Methods: Clinical examination, genome-wide linkage analysis, and exome sequencing were carried out on the family.
Results: Affected individuals presented with early-onset progressive mild hearing impairment with a fairly flat, gently downsloping or U-shaped audiogram configuration. Detailed clinical examination excluded any additional symptoms. Linkage analysis detected an interval on chromosome 1p21 with a logarithm of the odds (LOD) score of 8.29: designated locus DFNA37. Exome sequencing identified a novel canonical acceptor splice-site variant c.652-2A>C in the COL11A1 gene within the DFNA37 locus. Genotyping of all 48 family members confirmed segregation of this variant with the deafness phenotype in the extended family. The c.652-2A>C variant is novel, highly conserved, and confirmed in vitro to alter RNA splicing.
Conclusion: We have identified COL11A1 as the gene responsible for deafness at the DFNA37 locus. Previously, COL11A1 was solely associated with Marshall and Stickler syndromes. This study expands its phenotypic spectrum to include nonsyndromic deafness. The implications of this discovery are valuable in the clinical diagnosis, prognosis, and treatment of patients with COL11A1 pathogenic variants.
Keywords: COL11A1; DFNA37; exome sequencing; nonsyndromic hearing loss; splice-site variant.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
