

PIDD-dependent activation of caspase-2-mediated mitochondrial injury in E1A-induced cellular sensitivity to macrophage nitric oxide-induced apoptosis
- PMID: 30245858
- PMCID: PMC6135794
- DOI: 10.1038/s41420-018-0100-3
PIDD-dependent activation of caspase-2-mediated mitochondrial injury in E1A-induced cellular sensitivity to macrophage nitric oxide-induced apoptosis
Erratum in
-
Erratum: Publisher Correction: articles initially published in wrong volume.Cell Death Discov. 2019 Jul 10;5:116. doi: 10.1038/s41420-019-0186-2. eCollection 2019. Cell Death Discov. 2019. PMID: 31312525 Free PMC article.
Abstract
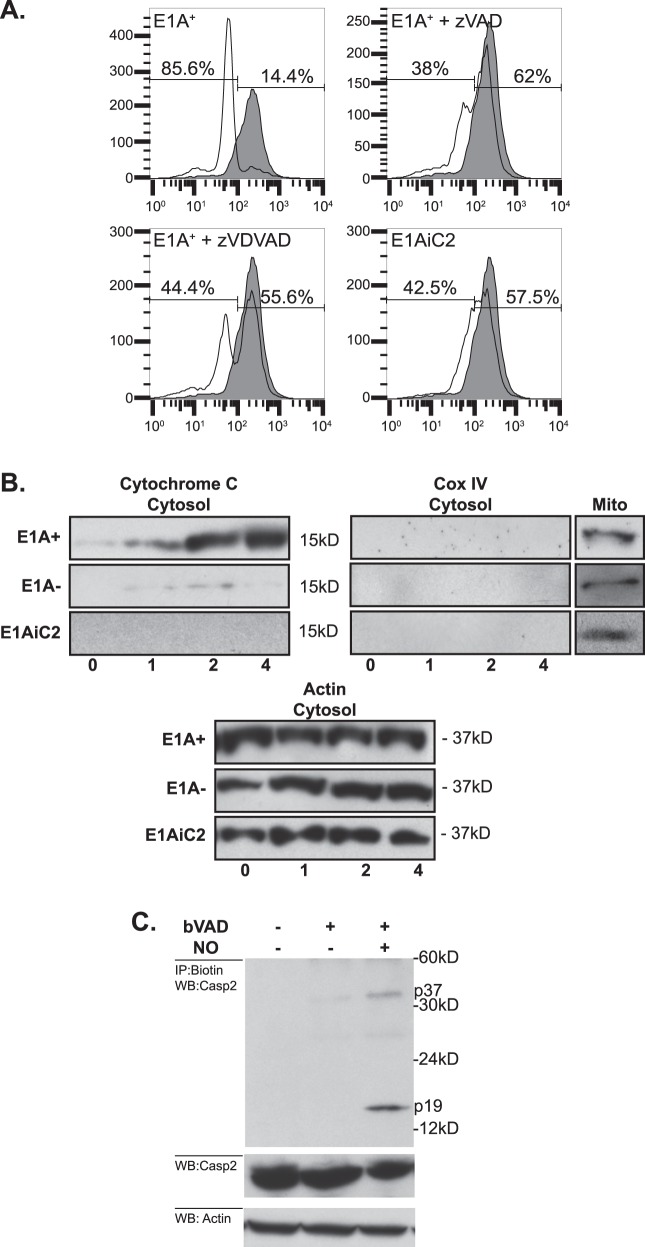
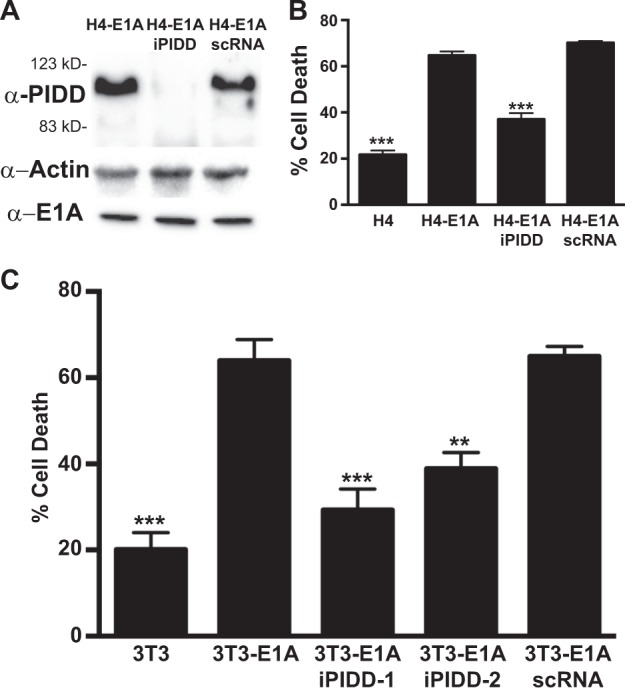
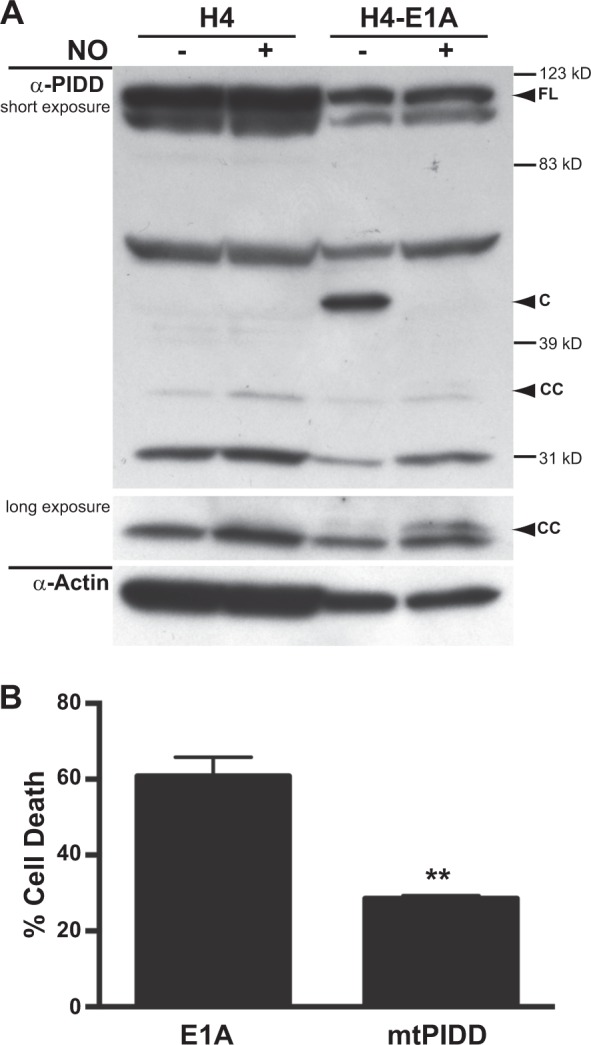
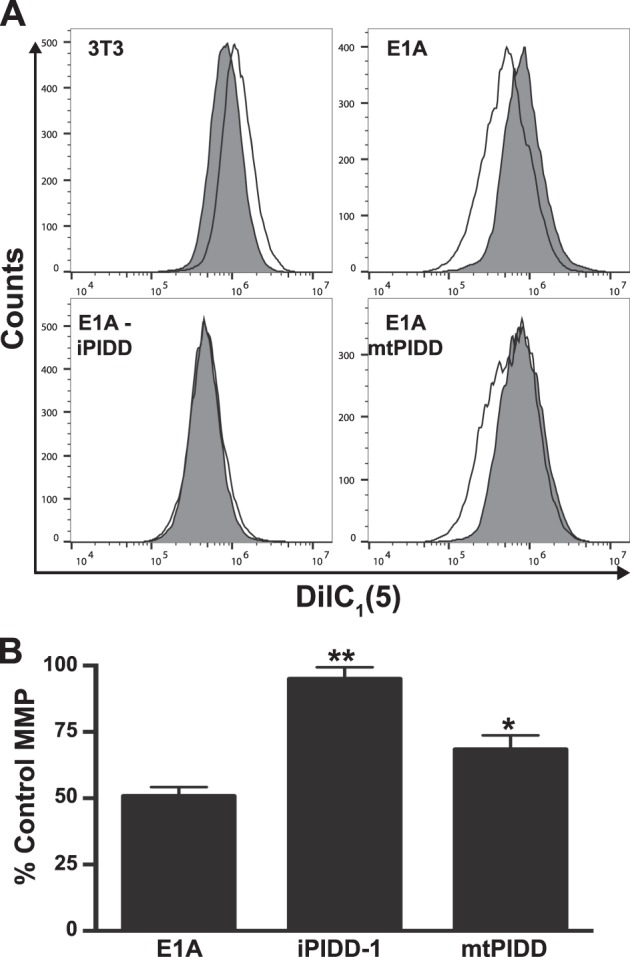
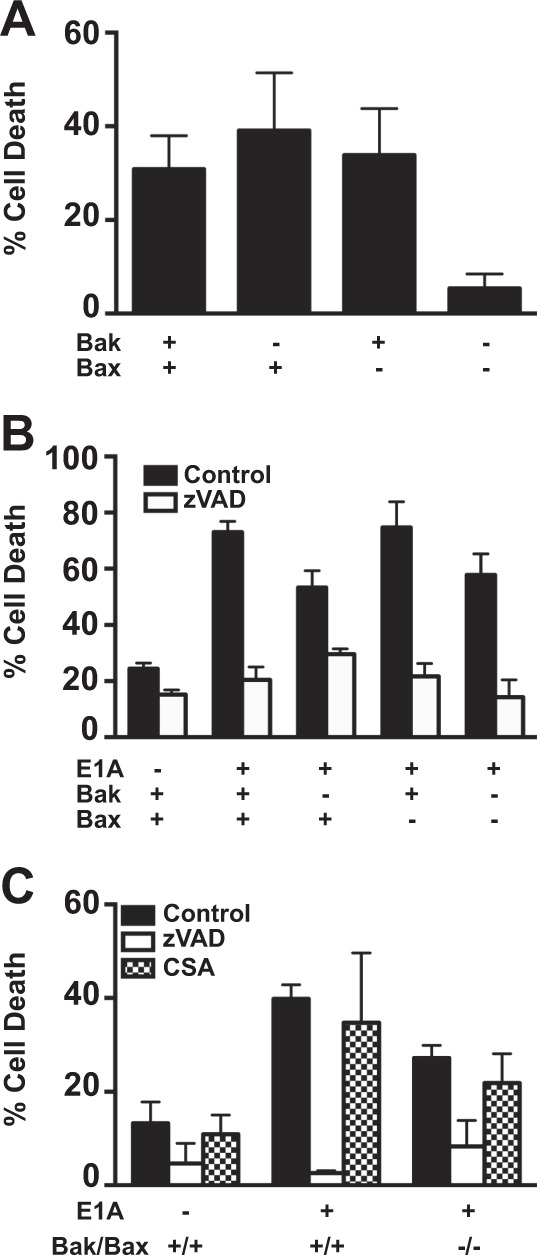
Expression of the adenovirus E1A oncogene sensitizes tumor cells to innate immune rejection by apoptosis induced by macrophage-produced tumor necrosis factor (TNF)-α and nitric oxide (NO). E1A sensitizes cells to TNF-α and NO through two distinct mechanisms, by repressing NF-κB-dependent antiapoptotic responses and enhancing caspase-2 activation and mitochondrial injury, respectively. The mechanisms through which E1A enhances caspase-2 activation in response to NO were unknown. Here, we report that E1A-induced sensitization to NO-induced apoptosis is dependent on expression of PIDD (p53-inducible protein with a death domain) and enhancement of primary immunodeficiency diseases (PIDD) processing for formation of the PIDDosome, the core component of the caspase-2 activation complex. NO-induced apoptosis in E1A-expressing cells did not require expression Bak or Bax, indicating that NO-induced caspase-2-mediated mitochondrial injury does not proceed through the activities of typical, proapoptotic Bcl-2 family members that induce mitochondrial cytochrome C release. These results define a PIDD-dependent pathway, through which E1A enhances casapse-2-mediated mitochondrial injury, resulting in increased sensitivity of mammalian cells to macrophage-induced, NO-mediated apoptosis.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Cook, J. L. & Routes, J. M. Role of the innate immune response in determining the tumorigenicity of neoplastic cells. Dev Biol 99–107 (2001) [discussion 107-08-143-60]. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
