

The APT complex is involved in non-coding RNA transcription and is distinct from CPF
- PMID: 30247719
- PMCID: PMC6265451
- DOI: 10.1093/nar/gky845
The APT complex is involved in non-coding RNA transcription and is distinct from CPF
Abstract
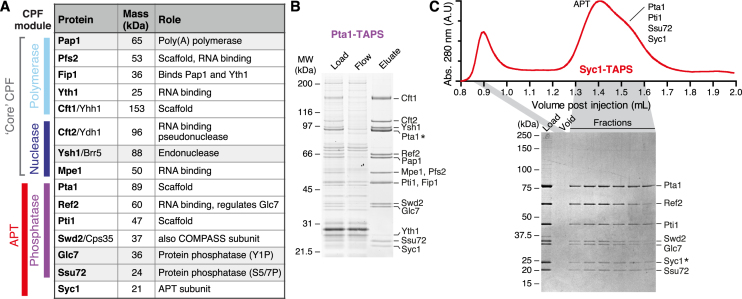
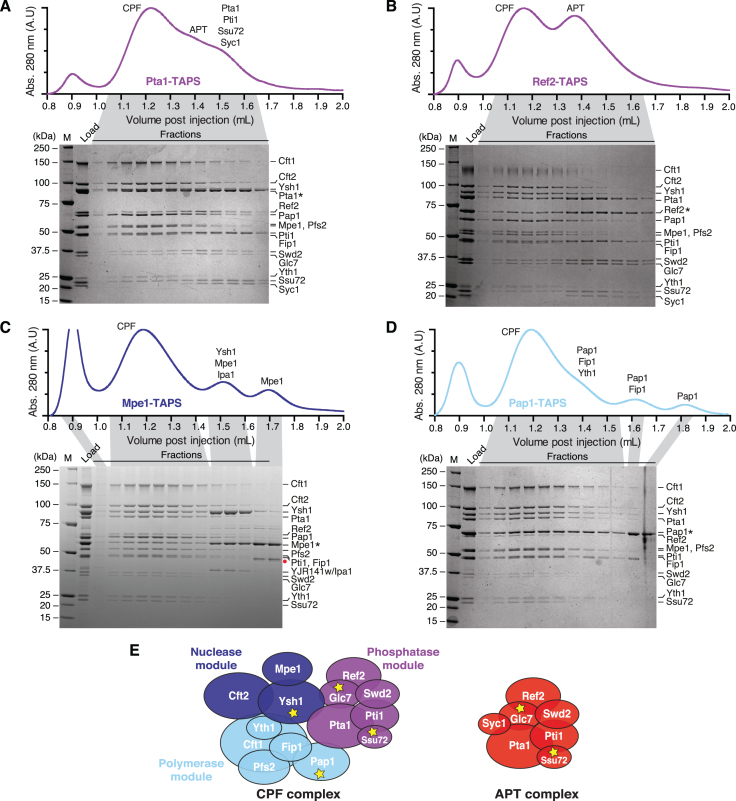
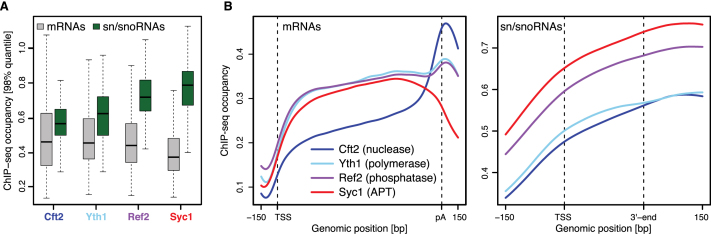
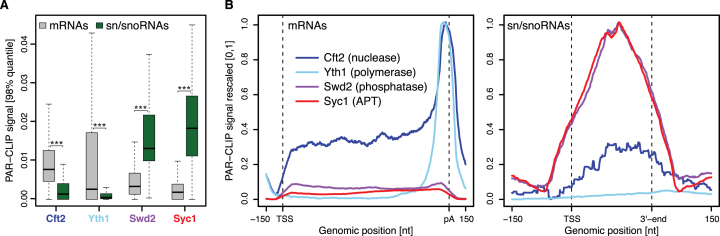
The 3'-ends of eukaryotic pre-mRNAs are processed in the nucleus by a large multiprotein complex, the cleavage and polyadenylation factor (CPF). CPF cleaves RNA, adds a poly(A) tail and signals transcription termination. CPF harbors four enzymatic activities essential for these processes, but how these are coordinated remains poorly understood. Several subunits of CPF, including two protein phosphatases, are also found in the related 'associated with Pta1' (APT) complex, but the relationship between CPF and APT is unclear. Here, we show that the APT complex is physically distinct from CPF. The 21 kDa Syc1 protein is associated only with APT, and not with CPF, and is therefore the defining subunit of APT. Using ChIP-seq, PAR-CLIP and RNA-seq, we show that Syc1/APT has distinct, but possibly overlapping, functions from those of CPF. Syc1/APT plays a more important role in sn/snoRNA production whereas CPF processes the 3'-ends of protein-coding pre-mRNAs. These results define distinct protein machineries for synthesis of mature eukaryotic protein-coding and non-coding RNAs.
Figures
References
-
- Jeronimo C., Collin P., Robert F.. The RNA polymerase II CTD: the Increasing complexity of a low-complexity protein domain. J. Mol. Biol. 2016; 428:2607–2622. - PubMed
-
- Gavin A.-C., Bösche M., Krause R., Grandi P., Marzioch M., Bauer A., Schultz J., Rick J.M., Michon A.-M., Cruciat C.-M. et al. . Functional organization of the yeast proteome by systematic analysis of protein complexes. Nature. 2002; 415:141–147. - PubMed
-
- Dichtl B., Blank D., Ohnacker M., Friedlein A., Roeder D., Langen H., Keller W.. A role for SSU72 in balancing RNA polymerase II transcription elongation and termination. Mol. Cell. 2002; 10:1139–1150. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
