

Gillespie syndrome in a South Asian child: a case report with confirmation of a heterozygous mutation of the ITPR1 gene and review of the clinical and molecular features
- PMID: 30249237
- PMCID: PMC6154888
- DOI: 10.1186/s12887-018-1286-5
Gillespie syndrome in a South Asian child: a case report with confirmation of a heterozygous mutation of the ITPR1 gene and review of the clinical and molecular features
Abstract
Background: Gillespie syndrome is a rare, congenital, neurological disorder characterized by the association of partial bilateral aniridia, non-progressive cerebellar ataxia and intellectual disability. Homozygous and heterozygous pathogenic variants of the ITPR1 gene encoding an inositol 1, 4, 5- triphosphate- responsive calcium channel have been identified in 13 patients recently. There have been 22 cases reported in the literature by 2016, mostly from the western hemisphere with none reported from Sri Lanka.
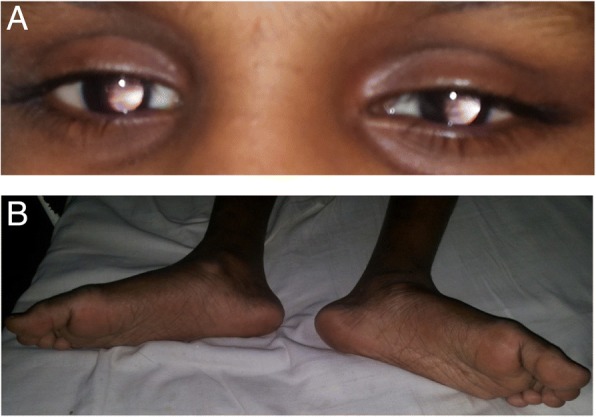
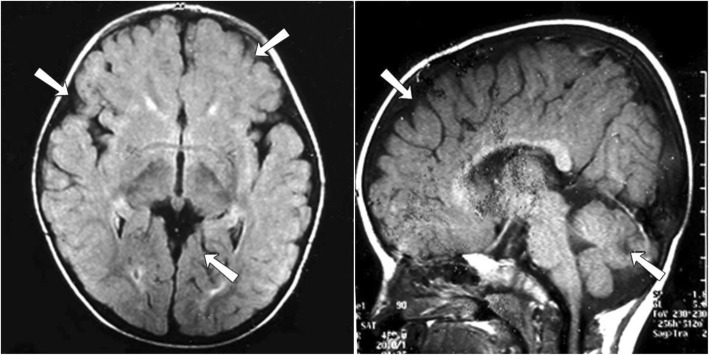
Case presentation: A 10-year-old girl born to healthy non-consanguineous parents with delayed development is described. She started walking unaided by 9 years with a significantly unsteady gait and her speech was similarly delayed. Physical examination revealed multiple cerebellar signs. Slit lamp examination of eyes revealed bilateral partial aniridia. Magnetic resonance imaging of brain at the age of 10 years revealed cerebellar (mainly vermian) hypoplasia. Genetic testing confirmed the clinical suspicion and demonstrated a heterozygous pathogenic variant c.7786_7788delAAG p.(Lys2596del) in the ITPR1 gene.
Conclusion: The report of this child with molecular confirmation of Gillespie syndrome highlights the need for careful evaluation of ophthalmological and neurological features in patients that enables correct clinical diagnosis. The availability of genetic testing enables more accurate counseling of the parents and patients regarding recurrence risks to other family members.
Keywords: Cerebellar hypoplasia; Gillespie syndrome; ITPR1 gene; Partial aniridia.
Conflict of interest statement
Ethics approval and consent to participate
Ethics approval was not sought as this patient was investigated as part of routine clinical care.
Written informed consent was obtained from the proband’s parents for genetic testing as part of standard care. A copy of the written consent is available for review by the corresponding author.
Consent for publication
Written informed consent was obtained from the proband’s parents for the publication of all personal information contained in this case report. A copy of the written consent is available for review by the corresponding author.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures

References
-
- Agarwal PK, Awan MA, Dutton GN, Strang N. Gillespie syndrome with impaired accommodation. J Pediatr Ophthalmol Strabismus. 2009;46(1):60. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
