

Inflammation: the link between comorbidities, genetics, and Alzheimer's disease
- PMID: 30249283
- PMCID: PMC6154824
- DOI: 10.1186/s12974-018-1313-3
Inflammation: the link between comorbidities, genetics, and Alzheimer's disease
Abstract
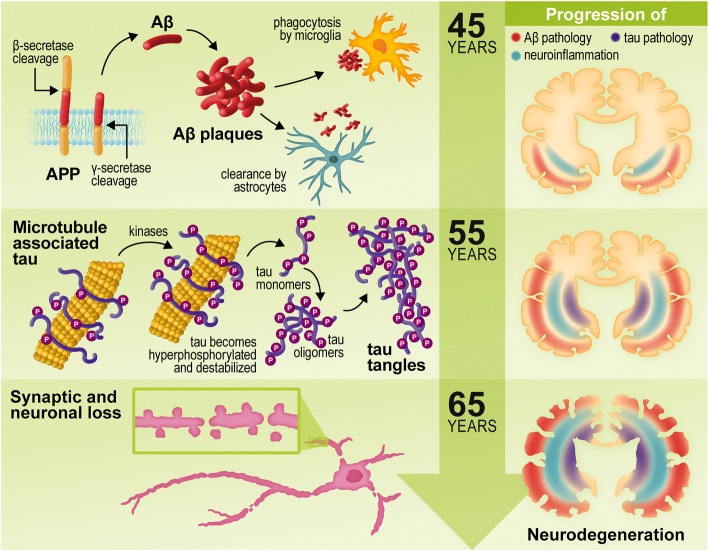
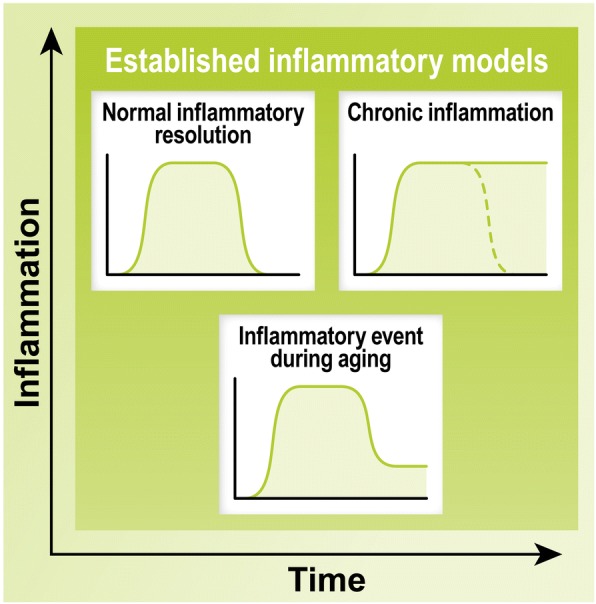
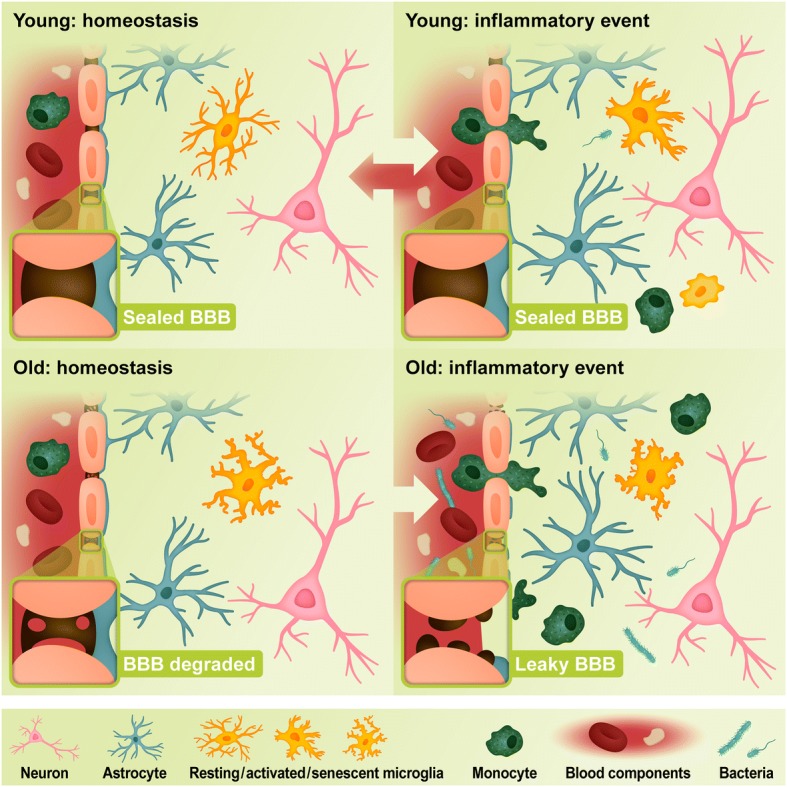
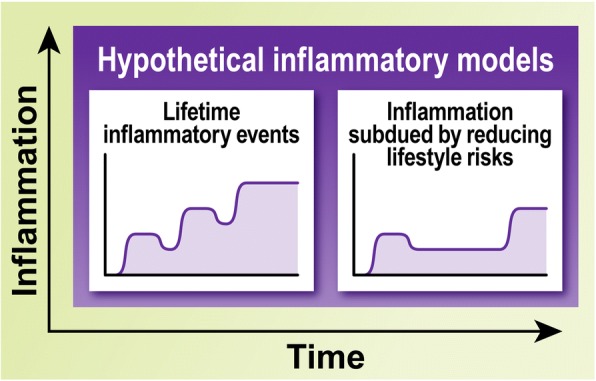
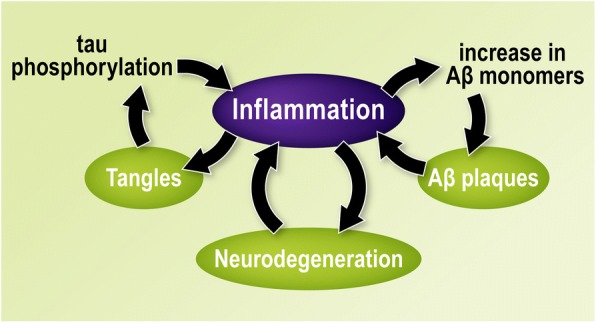
Alzheimer's disease (AD) is a neurodegenerative disorder, most cases of which lack a clear causative event. This has made the disease difficult to characterize and, thus, diagnose. Although some cases are genetically linked, there are many diseases and lifestyle factors that can lead to an increased risk of developing AD, including traumatic brain injury, diabetes, hypertension, obesity, and other metabolic syndromes, in addition to aging. Identifying common factors and trends between these conditions could enhance our understanding of AD and lead to the development of more effective treatments. Although the immune system is one of the body's key defense mechanisms, chronic inflammation has been increasingly linked with several age-related diseases. Moreover, it is now well accepted that chronic inflammation has an important role in the onset and progression of AD. In this review, the different inflammatory signals associated with AD and its risk factors will be outlined to demonstrate how chronic inflammation may be influencing individual susceptibility to AD. Our goal is to bring attention to potential shared signals presented by the immune system during different conditions that could lead to the development of successful treatments.
Keywords: APOE4; Aging; Alzheimer’s disease; CD33; Diabetes; Microglia; Neuroinflammation; Obesity; TBI; TREM2.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
