

Apoptosis and necroptosis in the liver: a matter of life and death
- PMID: 30250076
- PMCID: PMC6490680
- DOI: 10.1038/s41575-018-0065-y
Apoptosis and necroptosis in the liver: a matter of life and death
Abstract
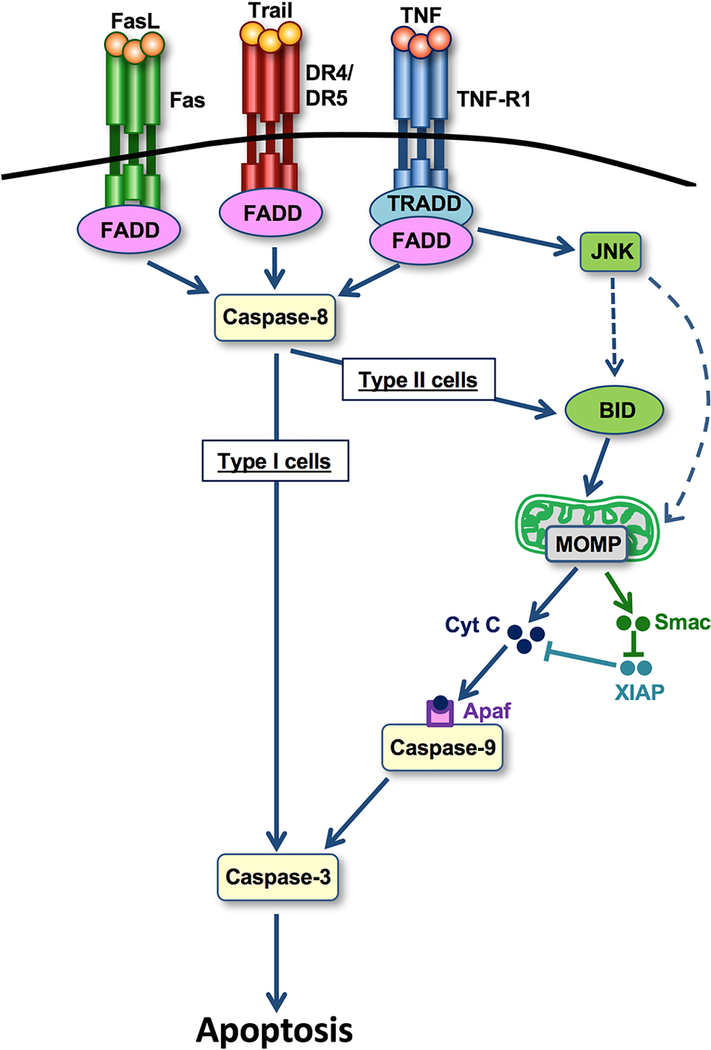
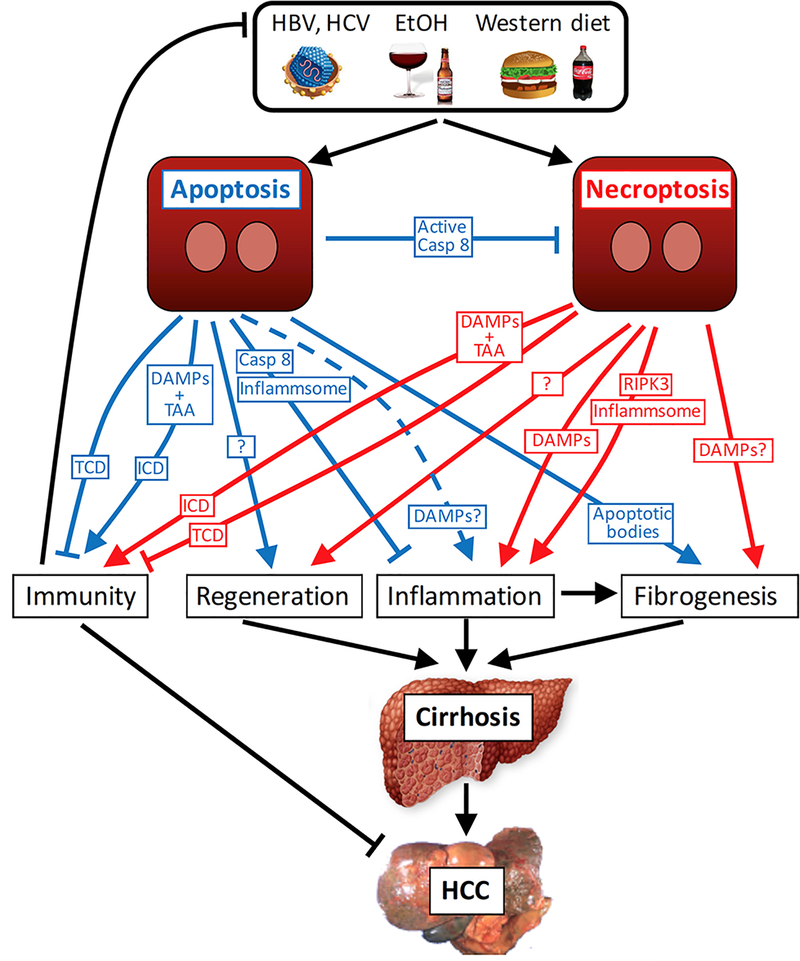
Cell death represents a basic biological paradigm that governs outcomes and long-term sequelae in almost every hepatic disease condition. Acute liver failure is characterized by massive loss of parenchymal cells but is usually followed by restitution ad integrum. By contrast, cell death in chronic liver diseases often occurs at a lesser extent but leads to long-term alterations in organ architecture and function, contributing to chronic hepatocyte turnover, the recruitment of immune cells and activation of hepatic stellate cells. These chronic cell death responses contribute to the development of liver fibrosis, cirrhosis and cancer. It has become evident that, besides apoptosis, necroptosis is a highly relevant form of programmed cell death in the liver. Differential activation of specific forms of programmed cell death might not only affect outcomes in liver diseases but also offer novel opportunities for therapeutic intervention. Here, we summarize the underlying molecular mechanisms and open questions about disease-specific activation and roles of programmed cell death forms, their contribution to response signatures and their detection. We focus on the role of apoptosis and necroptosis in acute liver injury, nonalcoholic fatty liver disease (NAFLD), nonalcoholic steatohepatitis (NASH) and liver cancer, and possible translations into clinical applications.
Figures

Similar articles
-
Lytic cell death in metabolic liver disease.J Hepatol. 2020 Aug;73(2):394-408. doi: 10.1016/j.jhep.2020.04.001. Epub 2020 Apr 13. J Hepatol. 2020. PMID: 32298766 Free PMC article. Review.
-
Mitochondrial Mechanisms of Apoptosis and Necroptosis in Liver Diseases.Anal Cell Pathol (Amst). 2021 Nov 11;2021:8900122. doi: 10.1155/2021/8900122. eCollection 2021. Anal Cell Pathol (Amst). 2021. PMID: 34804779 Free PMC article. Review.
-
Cell death and cell death responses in liver disease: mechanisms and clinical relevance.Gastroenterology. 2014 Oct;147(4):765-783.e4. doi: 10.1053/j.gastro.2014.07.018. Epub 2014 Jul 18. Gastroenterology. 2014. PMID: 25046161 Free PMC article. Review.
-
Loss of MLKL ameliorates liver fibrosis by inhibiting hepatocyte necroptosis and hepatic stellate cell activation.Theranostics. 2022 Jul 4;12(11):5220-5236. doi: 10.7150/thno.71400. eCollection 2022. Theranostics. 2022. PMID: 35836819 Free PMC article.
-
Decoding cell death signals in liver inflammation.J Hepatol. 2013 Sep;59(3):583-94. doi: 10.1016/j.jhep.2013.03.033. Epub 2013 Apr 6. J Hepatol. 2013. PMID: 23567086 Review.
Cited by
-
Mechanisms of nonalcoholic fatty liver disease and implications for surgery.Langenbecks Arch Surg. 2021 Feb;406(1):1-17. doi: 10.1007/s00423-020-01965-1. Epub 2020 Aug 24. Langenbecks Arch Surg. 2021. PMID: 32833053 Free PMC article. Review.
-
JNK signaling prevents biliary cyst formation through a CASPASE-8-dependent function of RIPK1 during aging.Proc Natl Acad Sci U S A. 2021 Mar 23;118(12):e2007194118. doi: 10.1073/pnas.2007194118. Proc Natl Acad Sci U S A. 2021. PMID: 33798093 Free PMC article.
-
Salidroside alleviates LPS-induced liver injury and inflammation through SIRT1- NF-κB pathway and NLRP3 inflammasome.Iran J Basic Med Sci. 2024;27(3):297-303. doi: 10.22038/IJBMS.2023.69401.15124. Iran J Basic Med Sci. 2024. PMID: 38333759 Free PMC article.
-
Infection-Associated Thymic Atrophy.Front Immunol. 2021 May 25;12:652538. doi: 10.3389/fimmu.2021.652538. eCollection 2021. Front Immunol. 2021. PMID: 34113341 Free PMC article. Review.
-
Sestrin2 maintains hepatic immune homeostasis and redox balance partially via inhibiting RIPK3-mediated necroptosis in metabolic dysfunction-associated steatohepatitis.Mol Metab. 2024 Feb;80:101865. doi: 10.1016/j.molmet.2023.101865. Epub 2023 Dec 30. Mol Metab. 2024. PMID: 38163459 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
