

Deciphering intra-species bacterial diversity of meat and seafood spoilage microbiota using gyrB amplicon sequencing: A comparative analysis with 16S rDNA V3-V4 amplicon sequencing
- PMID: 30252901
- PMCID: PMC6155546
- DOI: 10.1371/journal.pone.0204629
Deciphering intra-species bacterial diversity of meat and seafood spoilage microbiota using gyrB amplicon sequencing: A comparative analysis with 16S rDNA V3-V4 amplicon sequencing
Abstract
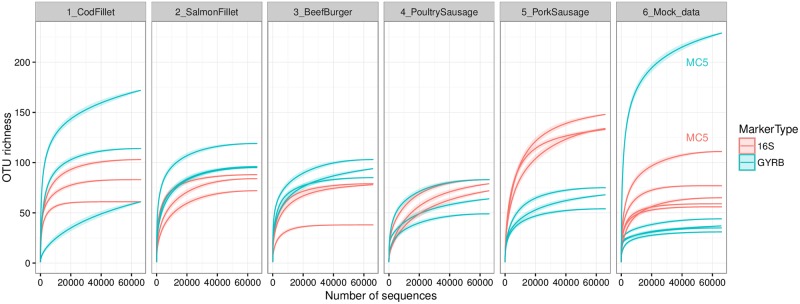
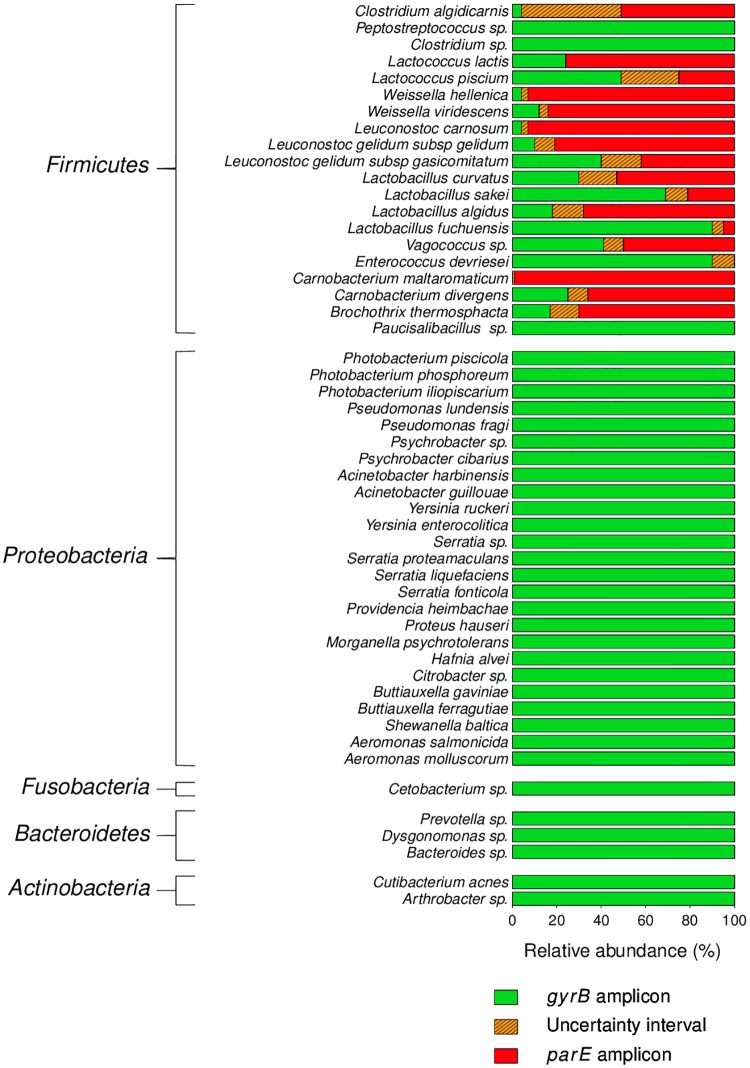
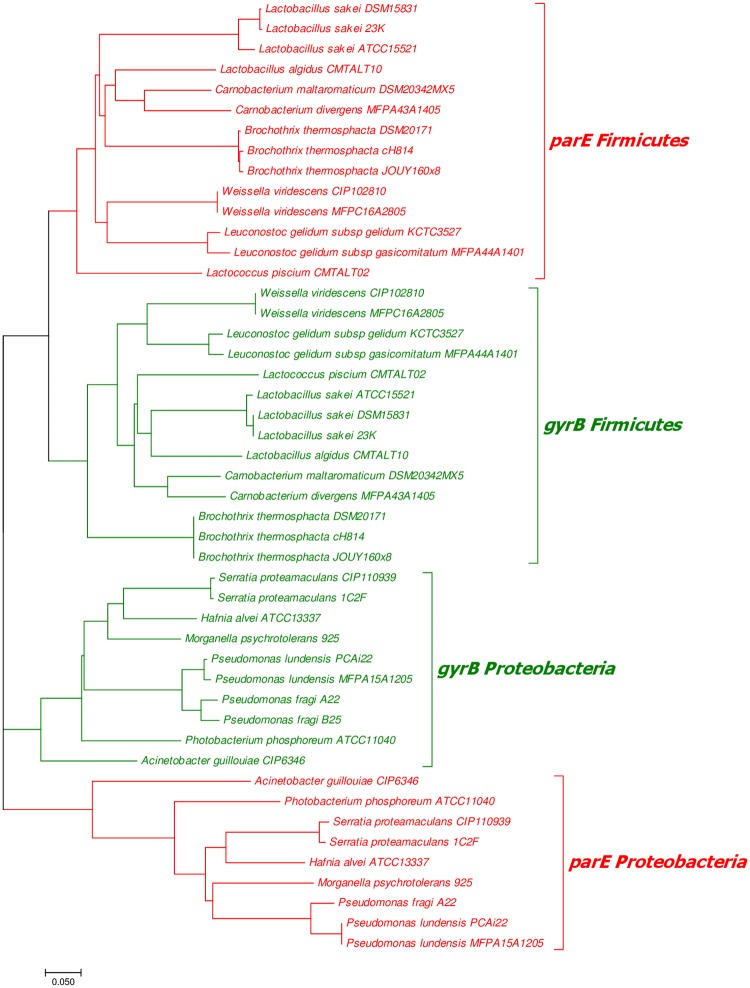
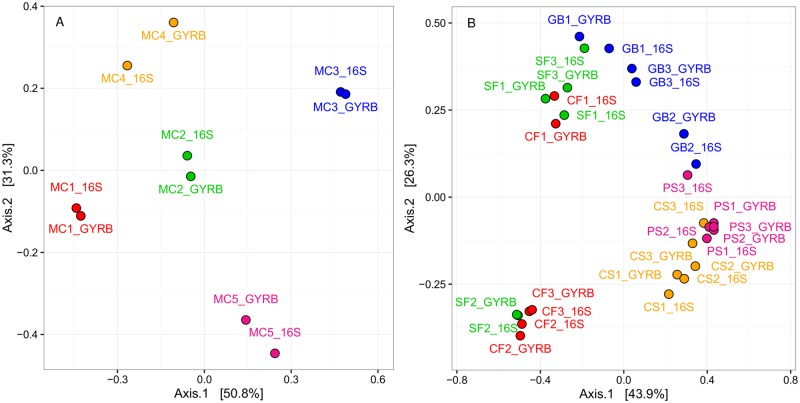
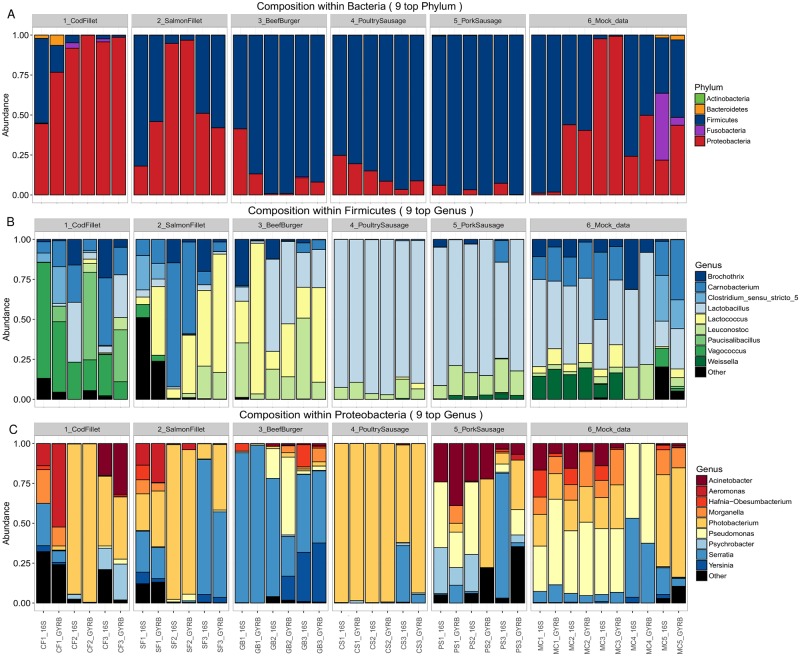
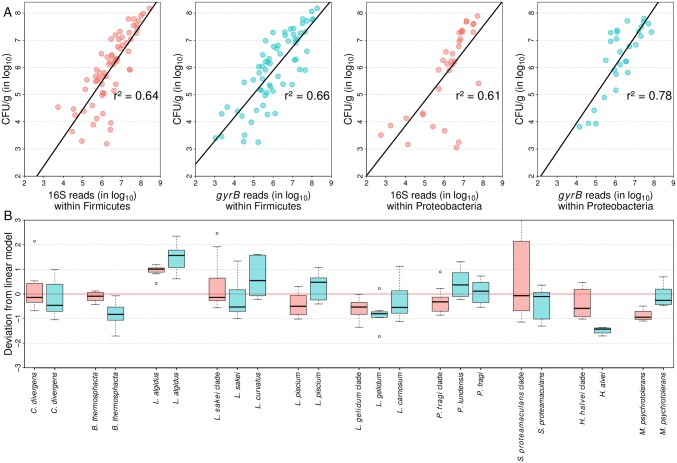
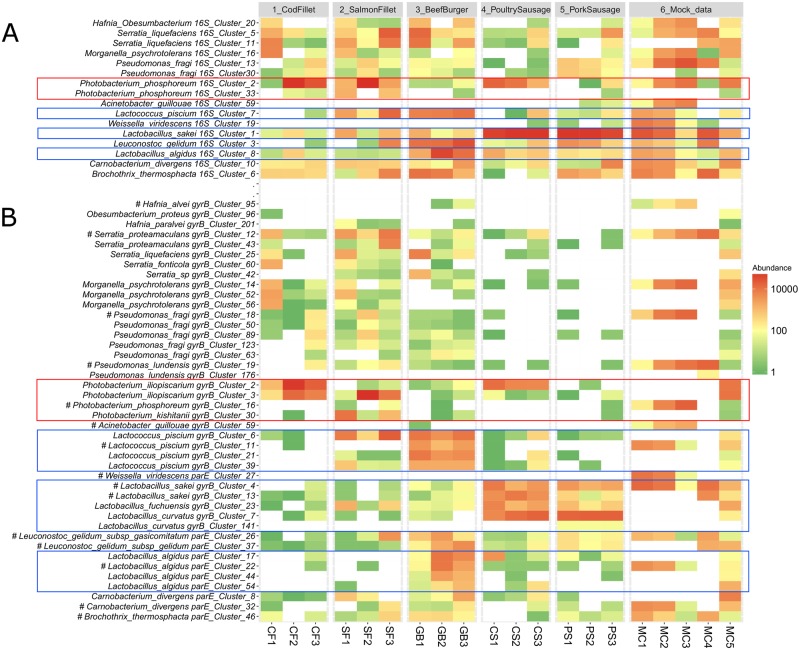
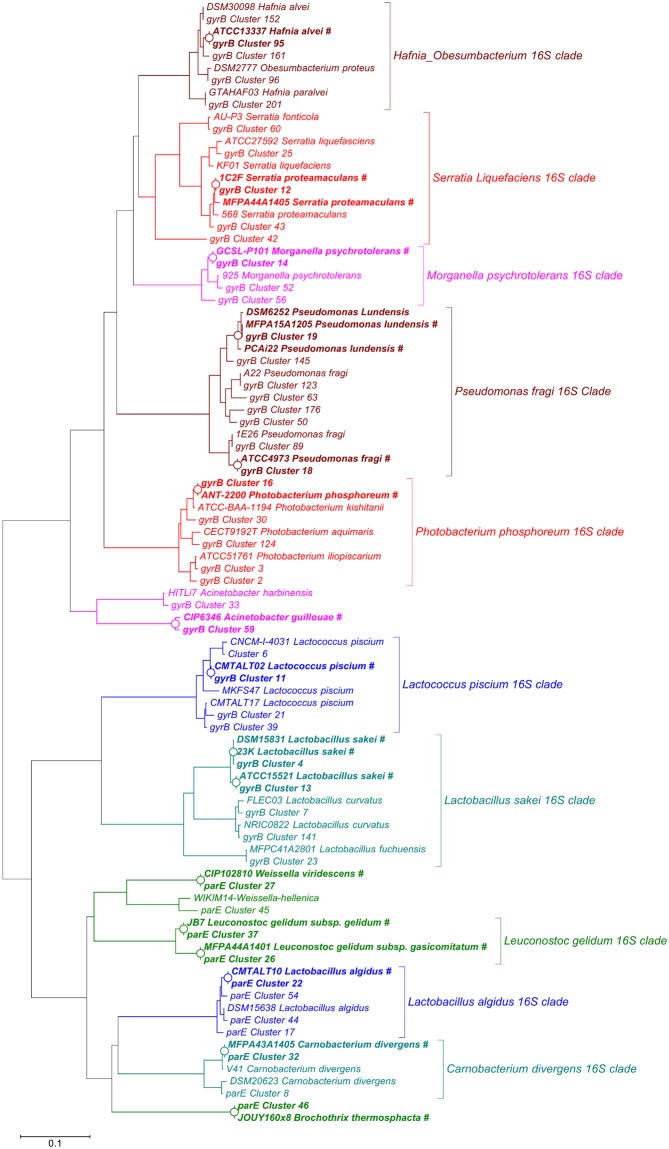
Meat and seafood spoilage ecosystems harbor extensive bacterial genomic diversity that is mainly found within a small number of species but within a large number of strains with different spoilage metabolic potential. To decipher the intraspecies diversity of such microbiota, traditional metagenetic analysis using the 16S rRNA gene is inadequate. We therefore assessed the potential benefit of an alternative genetic marker, gyrB, which encodes the subunit B of DNA gyrase, a type II DNA topoisomerase. A comparison between 16S rDNA-based (V3-V4) amplicon sequencing and gyrB-based amplicon sequencing was carried out in five types of meat and seafood products, with five mock communities serving as quality controls. Our results revealed that bacterial richness in these mock communities and food samples was estimated with higher accuracy using gyrB than using16S rDNA. However, for Firmicutes species, 35% of putative gyrB reads were actually identified as sequences of a gyrB paralog, parE, which encodes subunit B of topoisomerase IV; we therefore constructed a reference database of published sequences of both gyrB and pare for use in all subsequent analyses. Despite this co-amplification, the deviation between relative sequencing quantification and absolute qPCR quantification was comparable to that observed for 16S rDNA for all the tested species. This confirms that gyrB can be used successfully alongside 16S rDNA to determine the species composition (richness and evenness) of food microbiota. The major benefit of gyrB sequencing is its potential for improving taxonomic assignment and for further investigating OTU richness at the subspecies level, thus allowing more accurate discrimination of samples. Indeed, 80% of the reads of the 16S rDNA dataset were represented by thirteen 16S rDNA-based OTUs that could not be assigned at the species-level. Instead, these same clades corresponded to 44 gyrB-based OTUs, which differentiated various lineages down to the subspecies level. The increased ability of gyrB-based analyses to track and trace phylogenetically different groups of strains will generate improved resolution and more reliable results for studies of the strains implicated in food processes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
