

A Case Study of Genomic Instability in an Industrial Strain of Saccharomyces cerevisiae
- PMID: 30254181
- PMCID: PMC6222563
- DOI: 10.1534/g3.118.200446
A Case Study of Genomic Instability in an Industrial Strain of Saccharomyces cerevisiae
Abstract
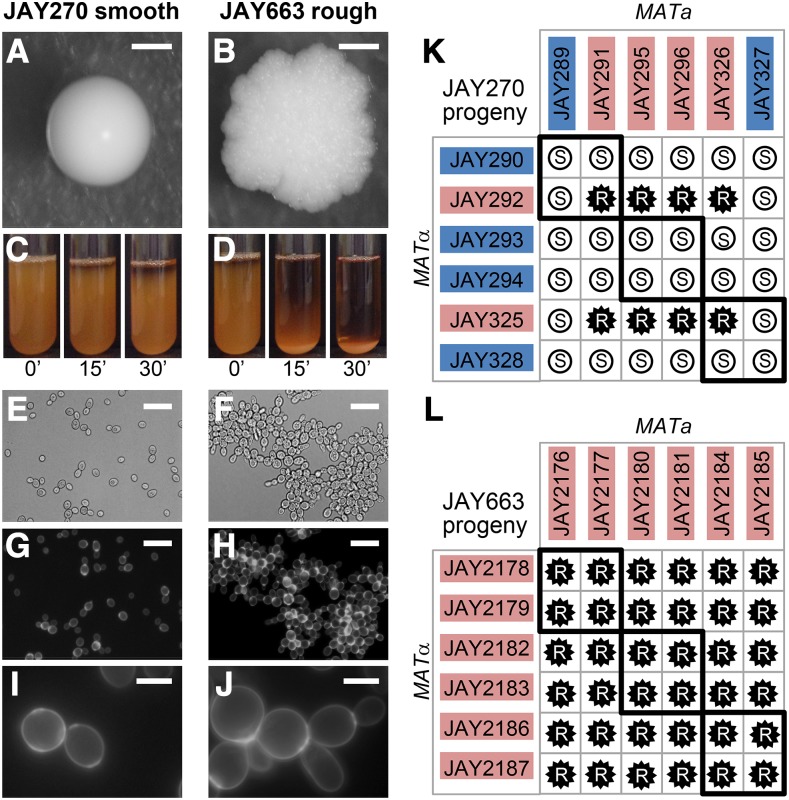
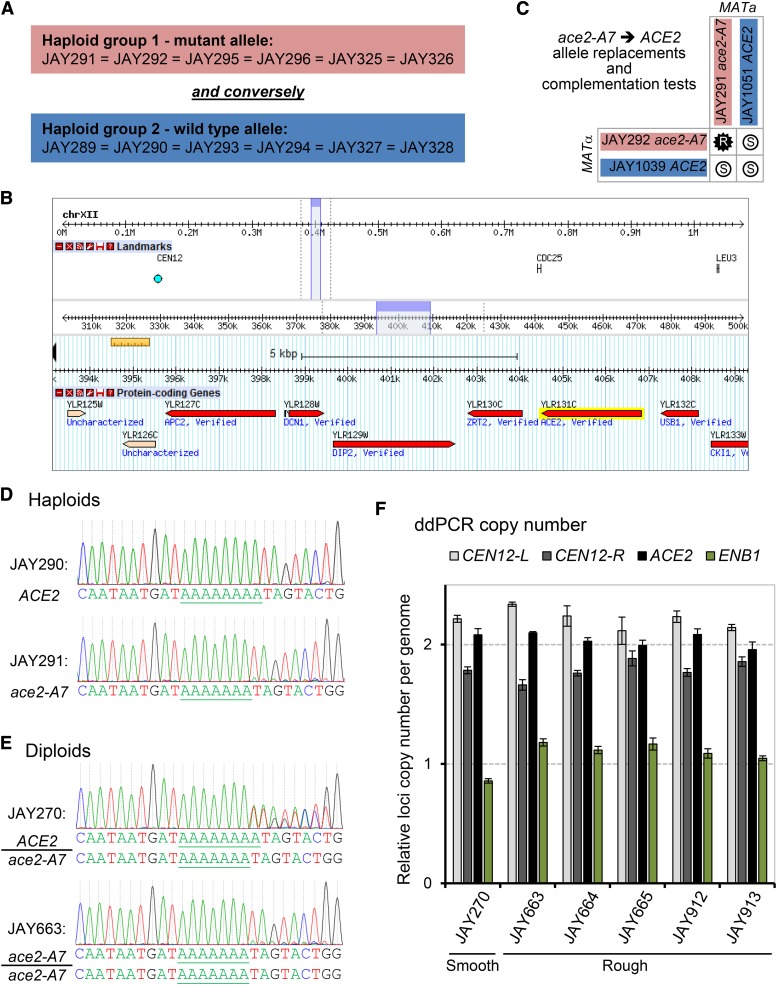
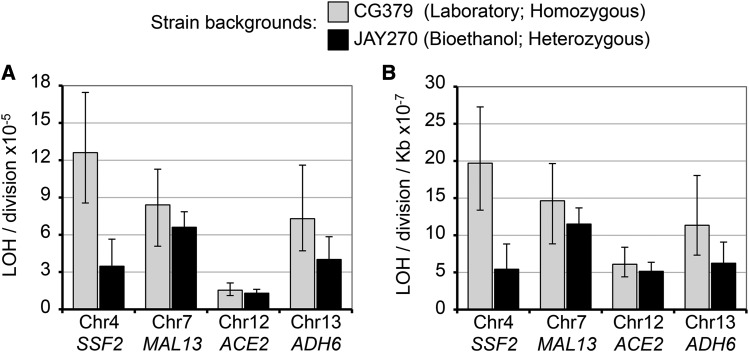
The Saccharomyces cerevisiae strain JAY270/PE2 is a highly efficient biocatalyst used in the production of bioethanol from sugarcane feedstock. This strain is heterothallic and diploid, and its genome is characterized by abundant structural and nucleotide polymorphisms between homologous chromosomes. One of the reasons it is favored by many distilleries is that its cells do not normally aggregate, a trait that facilitates cell recycling during batch-fed fermentations. However, long-term propagation makes the yeast population vulnerable to the effects of genomic instability, which may trigger the appearance of undesirable phenotypes such as cellular aggregation. In pure cultures of JAY270, we identified the recurrent appearance of mutants displaying a mother-daughter cell separation defect resulting in rough colonies in agar media and fast sedimentation in liquid culture. We investigated the genetic basis of the colony morphology phenotype and found that JAY270 is heterozygous for a frameshift mutation in the ACE2 gene (ACE2/ace2-A7), which encodes a transcriptional regulator of mother-daughter cell separation. All spontaneous rough colony JAY270-derived isolates analyzed carried copy-neutral loss-of-heterozygosity (LOH) at the region of chromosome XII where ACE2 is located (ace2-A7/ace2-A7). We specifically measured LOH rates at the ACE2 locus, and at three additional chromosomal regions in JAY270 and in a conventional homozygous diploid laboratory strain. This direct comparison showed that LOH rates at all sites were quite similar between the two strain backgrounds. In this case study of genomic instability in an industrial strain, we showed that the JAY270 genome is dynamic and that structural changes to its chromosomes can lead to new phenotypes. However, our analysis also indicated that the inherent level of genomic instability in this industrial strain is normal relative to a laboratory strain. Our work provides an important frame of reference to contextualize the interpretation of instability processes observed in the complex genomes of industrial yeast strains.
Keywords: ACE2; Colony morphology; Fermentation; Loss-of-heterozygosity; Mitotic recombination; Saccharomyces cerevisiae.
Copyright © by the Genetics Society of America.
Figures

Similar articles
-
Controlled Reduction of Genomic Heterozygosity in an Industrial Yeast Strain Reveals Wide Cryptic Phenotypic Variation.Front Genet. 2019 Sep 11;10:782. doi: 10.3389/fgene.2019.00782. eCollection 2019. Front Genet. 2019. PMID: 31572430 Free PMC article.
-
Genome structure of a Saccharomyces cerevisiae strain widely used in bioethanol production.Genome Res. 2009 Dec;19(12):2258-70. doi: 10.1101/gr.091777.109. Epub 2009 Oct 7. Genome Res. 2009. PMID: 19812109 Free PMC article.
-
Genome duplication and mutations in ACE2 cause multicellular, fast-sedimenting phenotypes in evolved Saccharomyces cerevisiae.Proc Natl Acad Sci U S A. 2013 Nov 5;110(45):E4223-31. doi: 10.1073/pnas.1305949110. Epub 2013 Oct 21. Proc Natl Acad Sci U S A. 2013. PMID: 24145419 Free PMC article.
-
The dynamics of loss of heterozygosity events in genomes.EMBO Rep. 2025 Feb;26(3):602-612. doi: 10.1038/s44319-024-00353-w. Epub 2025 Jan 2. EMBO Rep. 2025. PMID: 39747660 Free PMC article. Review.
-
Omics analysis of acetic acid tolerance in Saccharomyces cerevisiae.World J Microbiol Biotechnol. 2017 May;33(5):94. doi: 10.1007/s11274-017-2259-9. Epub 2017 Apr 12. World J Microbiol Biotechnol. 2017. PMID: 28405910 Review.
Cited by
-
Genome-Wide Analysis of Mitotic Recombination in Budding Yeast.Methods Mol Biol. 2021;2153:201-219. doi: 10.1007/978-1-0716-0644-5_15. Methods Mol Biol. 2021. PMID: 32840782 Free PMC article.
-
Punctuated Aneuploidization of the Budding Yeast Genome.Genetics. 2020 Sep;216(1):43-50. doi: 10.1534/genetics.120.303536. Epub 2020 Aug 4. Genetics. 2020. PMID: 32753390 Free PMC article.
-
Comparative Genomics Supports That Brazilian Bioethanol Saccharomyces cerevisiae Comprise a Unified Group of Domesticated Strains Related to Cachaça Spirit Yeasts.Front Microbiol. 2021 Apr 15;12:644089. doi: 10.3389/fmicb.2021.644089. eCollection 2021. Front Microbiol. 2021. PMID: 33936002 Free PMC article.
-
Optimal trade-off between boosted tolerance and growth fitness during adaptive evolution of yeast to ethanol shocks.Biotechnol Biofuels Bioprod. 2024 May 10;17(1):63. doi: 10.1186/s13068-024-02503-7. Biotechnol Biofuels Bioprod. 2024. PMID: 38730312 Free PMC article.
-
Controlled Reduction of Genomic Heterozygosity in an Industrial Yeast Strain Reveals Wide Cryptic Phenotypic Variation.Front Genet. 2019 Sep 11;10:782. doi: 10.3389/fgene.2019.00782. eCollection 2019. Front Genet. 2019. PMID: 31572430 Free PMC article.
References
-
- Ausubel F. M., Brent R., Kingston R. E., Moore D. D., Seidman J. G., et al. , 1998. Current Protocols in Molecular Biology, John Wiley & Sons, New York.
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases