

The Importance of an Early and Accurate MEN1 Diagnosis
- PMID: 30254610
- PMCID: PMC6141626
- DOI: 10.3389/fendo.2018.00533
The Importance of an Early and Accurate MEN1 Diagnosis
Abstract
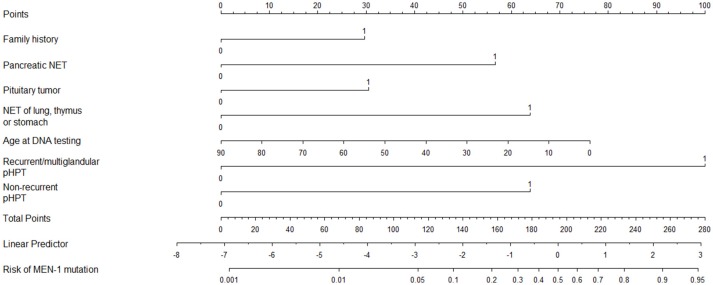
Multiple Endocrine Neoplasia type 1 (MEN1) is a rare autosomal dominant inherited condition, causing significant morbidity, and a reduction of life expectancy. A timely and accurate diagnosis of MEN1 is paramount to improve disease outcomes. This enables early identification of tumor manifestations allowing timely treatment for reducing morbidity and improving survival. Current management of MEN1 poses two challenges regarding the MEN1 diagnosis: diagnostic delay and the issue of phenocopies. A delay in diagnosis can be caused by a delay in identifying the index case, and by a delay in identifying affected family members of an index case. At present, lag time between diagnosis of MEN1 in index cases and genetic testing of family members was estimated to be 3.5 years. A subsequent delay in diagnosing affected family members was demonstrated to cause potential harm. Non-index cases have been found to develop clinically relevant tumor manifestations during the lag times. Centralized care, monitoring of patients outcomes on a national level and thereby improving awareness of physicians treating MEN1 patients, will contribute to improved care. The second challenge relates to "phenocopies." Phenocopies refers to the 5-25% of clinically diagnosed patients with MEN1in whom no mutation can be found. Up to now, the clinical diagnosis of MEN1 is defined as the simultaneous presence of at least two of the three characteristic tumors (pituitary, parathyroids, or pancreatic islets). These clinically diagnosed patients undergo intensive follow up. Recent insights, however, challenge the validity of this clinical criterion. The most common mutation-negative MEN1 phenotype is the combination of primary hyperparathyroidism and a pituitary adenoma. This phenotype might also be caused by mutations in the CDKN1B gene, causing the recently described MEN4 syndrome. Moreover, primary hyperparathyroidism and pituitary adenoma are relatively common in the general population. Limiting follow-up in patients with a sporadic co-occurrence of pHPT and PIT could reduce exposure to radiation from imaging, healthcare costs and anxiety.
Keywords: MEN1; delayed diagnosis; diagnosis; epidemiology; genetic testing.
Figures
References
-
- Pieterman CR, van Hulsteijn LT, den HM, van der Luijt RB, Bonenkamp JJ, Hermus AR, et al. Primary hyperparathyroidism in MEN1 patients: a cohort study with longterm follow-up on preferred surgical procedure and the relation with genotype. Ann Surg. (2012) 255:1171–8. 10.1097/SLA.0b013e31824c5145 - DOI - PubMed
-
- Pieterman CRC, Conemans EB, Dreijerink KMA, de Laat JM, Timmers HTM, Vriens MR, et al. Thoracic and duodenopancreatic neuroendocrine tumors in multiple endocrine neoplasia type 1: natural history and function of menin in tumorigenesis. Endocr Relat Cancer (2014) 21:R121–42. 10.1530/ERC-13-0482 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous