

Proteinuric Kidney Diseases: A Podocyte's Slit Diaphragm and Cytoskeleton Approach
- PMID: 30255020
- PMCID: PMC6141722
- DOI: 10.3389/fmed.2018.00221
Proteinuric Kidney Diseases: A Podocyte's Slit Diaphragm and Cytoskeleton Approach
Abstract
Proteinuric kidney diseases are a group of disorders with diverse pathological mechanisms associated with significant losses of protein in the urine. The glomerular filtration barrier (GFB), comprised of the three important layers, the fenestrated glomerular endothelium, the glomerular basement membrane (GBM), and the podocyte, dictates that disruption of any one of these structures should lead to proteinuric disease. Podocytes, in particular, have long been considered as the final gatekeeper of the GFB. This specialized visceral epithelial cell contains a complex framework of cytoskeletons forming foot processes and mediate important cell signaling to maintain podocyte health. In this review, we will focus on slit diaphragm proteins such as nephrin, podocin, TRPC6/5, as well as cytoskeletal proteins Rho/small GTPases and synaptopodin and their respective roles in participating in the pathogenesis of proteinuric kidney diseases. Furthermore, we will summarize the potential therapeutic options targeting the podocyte to treat this group of kidney diseases.
Keywords: Rho/small GTPases; TRPC5/6; cytoskeleton; nephrin; podocin; podocyte; slit diaphragm; synaptopodin.
Figures
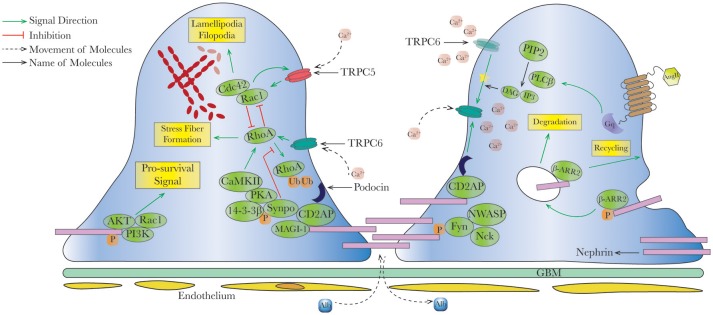
 ). Endocytosis and recycling of proteins on slit diaphragm is controlled by serial phosphorylation and associated proteins such as β-arrestin2. PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; Rac1, ras-related C3 botulinum toxin substrate 1; Cdc42, cell division control protein 42 homolog; RhoA, ras homolog gene family, member A; CaMKII, ca2+/calmodulin-dependent protein kinase II; Synpo, synaptopodin; MAGI-1, membrane-associated guanylate kinase inverted-1; PIP2, phosphatidylinositol bisphosphate; PLCβ, phospholipase Cβ; DAG, diacylglycerol; IP3, inositol 1,4,5-trisphosphate; β-ARR2, β-arrestin 2.
). Endocytosis and recycling of proteins on slit diaphragm is controlled by serial phosphorylation and associated proteins such as β-arrestin2. PI3K, phosphatidylinositol-4,5-bisphosphate 3-kinase; Rac1, ras-related C3 botulinum toxin substrate 1; Cdc42, cell division control protein 42 homolog; RhoA, ras homolog gene family, member A; CaMKII, ca2+/calmodulin-dependent protein kinase II; Synpo, synaptopodin; MAGI-1, membrane-associated guanylate kinase inverted-1; PIP2, phosphatidylinositol bisphosphate; PLCβ, phospholipase Cβ; DAG, diacylglycerol; IP3, inositol 1,4,5-trisphosphate; β-ARR2, β-arrestin 2.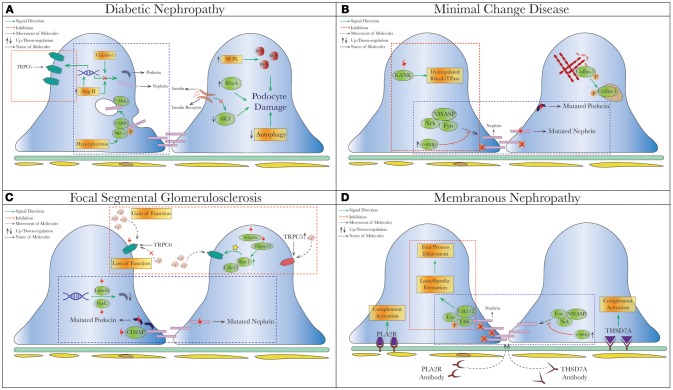
 (dysregulated pathways related to SD proteins) or red
(dysregulated pathways related to SD proteins) or red  (cytoskeletal dysregulation via TRPC or Rho-GTPase) dashed square. (A) Diabetic Nephropathy. Under hyperglycemia, nephrin endocytosis can be induced by the interaction of β-arrestin2 with PKC-α (59, 60). Increased AngII (78, 158) and Claudin-1 (159, 203) in diabetes were both shown to reduce expression of nephrin and podocin. On the contrary, increased TRPC6 expression mediated by AngII was seen in streptozotocin-induced diabetic rats causing podocytopenia and proteinuria (129, 130). Hyperglycemia also leads to podocyte apoptosis from production of reactive oxygen species (ROS) through NAPDH oxidase (NOX) (204), so as does insulin resistance due to reduced AKT pro-survival signaling (205). (B) Minimal Change Disease. Mutations of nephrin (NPHS1) and podocin (NPHS2) have been reported in MCD and tended to have higher rates of steroid-resistance (119, 174, 206), as well as recessive mutations of KANK (kidney ankyrin repeating-containing protein) KANK1 and KANK2 identified in a cohort of Arab and European origins (187). Cofilin-1, actin-binding protein necessary for maintaining podocyte architecture (207), is inactivated by phosphorylation seen in human MCD, leading to its redistribution to nucleus in the disease states (188). A novel molecule c-mip noted to be upregulated in human MN/MCD was shown to impair podocyte actin reorganization by inhibiting interaction between Fyn/N-WASP and nephrin/Nck. Podocyte overexpressed with c-mip could result in downregulation of nephrin and synaptopodin (52, 185, 208). (C) Focal Segmental Glomerulosclerosis (FSGS). Multiple gene mutations have been identified in patients with FSGS through genetic studies. Most of them encode various critical podocyte structures or signaling pathways, such as SD complex (NPHS1, NPHS2, CD2AP), SD-related Ca2+ signaling (TRPC6, PLCE1), actin cytoskeleton/endocytosis (ACTN4, INF2, FAT1, MYO1E), and small-GTPases (AHRGP24, ARGHDIA, ARHGEF17) (–214). A novel mechanism of increased podocin endocytosis via sorting nexin 9 (SNX9), which is seen in human IgA nephropathy, membranous nephropathy and FSGS was also described (171). Blocking the imbalanced TRPC5/6 signaling and increased circulating permeability factors (suPAR/CLC-1) are possible new therapeutic approaches in FSGS. (D) Membranous Nephropathy. Besides the well-known antibody-mediated primary MN (anti-PLA2R/THSd7A), reduced nephrin phosphorylation (170) and increased podocin endocytosis (171) have been described in human MN. Disorganization of actin cytoskeleton from a different cytoskeletal pathway Cas-FAK-Crk1/2 activation (not the typical nephrin-Nck pathway) was also seen in human MCD and MN (186). Mutation (red thunder
(cytoskeletal dysregulation via TRPC or Rho-GTPase) dashed square. (A) Diabetic Nephropathy. Under hyperglycemia, nephrin endocytosis can be induced by the interaction of β-arrestin2 with PKC-α (59, 60). Increased AngII (78, 158) and Claudin-1 (159, 203) in diabetes were both shown to reduce expression of nephrin and podocin. On the contrary, increased TRPC6 expression mediated by AngII was seen in streptozotocin-induced diabetic rats causing podocytopenia and proteinuria (129, 130). Hyperglycemia also leads to podocyte apoptosis from production of reactive oxygen species (ROS) through NAPDH oxidase (NOX) (204), so as does insulin resistance due to reduced AKT pro-survival signaling (205). (B) Minimal Change Disease. Mutations of nephrin (NPHS1) and podocin (NPHS2) have been reported in MCD and tended to have higher rates of steroid-resistance (119, 174, 206), as well as recessive mutations of KANK (kidney ankyrin repeating-containing protein) KANK1 and KANK2 identified in a cohort of Arab and European origins (187). Cofilin-1, actin-binding protein necessary for maintaining podocyte architecture (207), is inactivated by phosphorylation seen in human MCD, leading to its redistribution to nucleus in the disease states (188). A novel molecule c-mip noted to be upregulated in human MN/MCD was shown to impair podocyte actin reorganization by inhibiting interaction between Fyn/N-WASP and nephrin/Nck. Podocyte overexpressed with c-mip could result in downregulation of nephrin and synaptopodin (52, 185, 208). (C) Focal Segmental Glomerulosclerosis (FSGS). Multiple gene mutations have been identified in patients with FSGS through genetic studies. Most of them encode various critical podocyte structures or signaling pathways, such as SD complex (NPHS1, NPHS2, CD2AP), SD-related Ca2+ signaling (TRPC6, PLCE1), actin cytoskeleton/endocytosis (ACTN4, INF2, FAT1, MYO1E), and small-GTPases (AHRGP24, ARGHDIA, ARHGEF17) (–214). A novel mechanism of increased podocin endocytosis via sorting nexin 9 (SNX9), which is seen in human IgA nephropathy, membranous nephropathy and FSGS was also described (171). Blocking the imbalanced TRPC5/6 signaling and increased circulating permeability factors (suPAR/CLC-1) are possible new therapeutic approaches in FSGS. (D) Membranous Nephropathy. Besides the well-known antibody-mediated primary MN (anti-PLA2R/THSd7A), reduced nephrin phosphorylation (170) and increased podocin endocytosis (171) have been described in human MN. Disorganization of actin cytoskeleton from a different cytoskeletal pathway Cas-FAK-Crk1/2 activation (not the typical nephrin-Nck pathway) was also seen in human MCD and MN (186). Mutation (red thunder  ) of proteins, inhibiting (red cross
) of proteins, inhibiting (red cross ) or activating (yellow star) different signal pathways leads to reduced level of phosphorylation, increased intracellular calcium and endocytic process. AngII, Angiotensin II; c-ABL, Abelson murine leukemia viral oncogene homolog 1; RhoA, ras homolog gene family, member A; NOX, NAPDH oxidase; ROS, reactive oxygen species; Cdc42, cell division control protein 42 homolog; Rac1, ras-related C3 botulinum toxin substrate 1; Lmx1b, LIM homeobox transcription factor 1-beta; SNX-9, sorting nexin 9; suPAR, soluble urokinase plasminogen activator receptor; CLC-1, cardiotropin-like cytokine-1; FAK, focal adhesion kinase; PLA2R, phospholipase A2 receptor; THSD7A, thrombospondin Type 1 domain containing 7A; c-mip, c-maf-inducing protein; KANK, kidney ankyrin repeating-containing protein.
) or activating (yellow star) different signal pathways leads to reduced level of phosphorylation, increased intracellular calcium and endocytic process. AngII, Angiotensin II; c-ABL, Abelson murine leukemia viral oncogene homolog 1; RhoA, ras homolog gene family, member A; NOX, NAPDH oxidase; ROS, reactive oxygen species; Cdc42, cell division control protein 42 homolog; Rac1, ras-related C3 botulinum toxin substrate 1; Lmx1b, LIM homeobox transcription factor 1-beta; SNX-9, sorting nexin 9; suPAR, soluble urokinase plasminogen activator receptor; CLC-1, cardiotropin-like cytokine-1; FAK, focal adhesion kinase; PLA2R, phospholipase A2 receptor; THSD7A, thrombospondin Type 1 domain containing 7A; c-mip, c-maf-inducing protein; KANK, kidney ankyrin repeating-containing protein.References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources