

De Novo Sequence and Copy Number Variants Are Strongly Associated with Tourette Disorder and Implicate Cell Polarity in Pathogenesis
- PMID: 30257206
- PMCID: PMC6475626
- DOI: 10.1016/j.celrep.2018.08.082
De Novo Sequence and Copy Number Variants Are Strongly Associated with Tourette Disorder and Implicate Cell Polarity in Pathogenesis
Erratum in
-
De Novo Sequence and Copy Number Variants Are Strongly Associated with Tourette Disorder and Implicate Cell Polarity in Pathogenesis.Cell Rep. 2018 Dec 18;25(12):3544. doi: 10.1016/j.celrep.2018.12.024. Cell Rep. 2018. PMID: 30566877 No abstract available.
Abstract
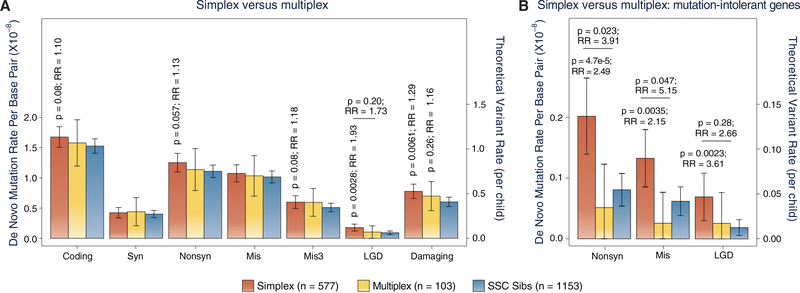
We previously established the contribution of de novo damaging sequence variants to Tourette disorder (TD) through whole-exome sequencing of 511 trios. Here, we sequence an additional 291 TD trios and analyze the combined set of 802 trios. We observe an overrepresentation of de novo damaging variants in simplex, but not multiplex, families; we identify a high-confidence TD risk gene, CELSR3 (cadherin EGF LAG seven-pass G-type receptor 3); we find that the genes mutated in TD patients are enriched for those related to cell polarity, suggesting a common pathway underlying pathobiology; and we confirm a statistically significant excess of de novo copy number variants in TD. Finally, we identify significant overlap of de novo sequence variants between TD and obsessive-compulsive disorder and de novo copy number variants between TD and autism spectrum disorder, consistent with shared genetic risk.
Keywords: TIC Genetics; Tourette disorder; cell polarity; copy number variants; de novo variants; gene discovery; microarray genotyping; multiplex; simplex; whole exome sequencing.
Copyright © 2018 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
Donald L. Gilbert has received salary/travel/honoraria from the Tourette Association of America, the Child Neurology Society, U.S. National Vaccine Injury Compensation Program, Ecopipam Pharmaceuticals, EryDel Pharmaceuticals, Elsevier, and Wolters Kluwer. A.J.W. is a paid consultant for Daiichi Sankyo. M.W.S. is a consultant to BlackThorn and ArRett Pharmaceuticals.
Figures


References
-
- Bel C, Oguievetskaia K, Pitaval C, Goutebroze L, and Faivre-Sarrailh C (2009). Axonal targeting of Caspr2 in hippocampal neurons via selective somatodendritic endocytosis. J. Cell Sci 122, 3403–3413. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- R01 MH092292/MH/NIMH NIH HHS/United States
- R01 NS102371/NS/NINDS NIH HHS/United States
- R01 MH092513/MH/NIMH NIH HHS/United States
- R01 MH115993/MH/NIMH NIH HHS/United States
- R01 MH115958/MH/NIMH NIH HHS/United States
- K08 MH099424/MH/NIMH NIH HHS/United States
- R01 MH092520/MH/NIMH NIH HHS/United States
- R01 MH092289/MH/NIMH NIH HHS/United States
- U01 GM115486/GM/NIGMS NIH HHS/United States
- R01 MH115959/MH/NIMH NIH HHS/United States
- UL1 TR001102/TR/NCATS NIH HHS/United States
- R01 MH092293/MH/NIMH NIH HHS/United States
- U24 HG008956/HG/NHGRI NIH HHS/United States
- UM1 HG006504/HG/NHGRI NIH HHS/United States
- R01 MH092516/MH/NIMH NIH HHS/United States
- R01 NS105746/NS/NINDS NIH HHS/United States
- R01 MH092291/MH/NIMH NIH HHS/United States
- R25 MH077823/MH/NIMH NIH HHS/United States
- R01 MH092290/MH/NIMH NIH HHS/United States
- R01 GM115486/GM/NIGMS NIH HHS/United States
- R01 MH115962/MH/NIMH NIH HHS/United States
- K02 NS085048/NS/NINDS NIH HHS/United States
- R01 MH115961/MH/NIMH NIH HHS/United States
- U24 MH068457/MH/NIMH NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
