

Synthesizing Signaling Pathways from Temporal Phosphoproteomic Data
- PMID: 30257219
- PMCID: PMC6295338
- DOI: 10.1016/j.celrep.2018.08.085
Synthesizing Signaling Pathways from Temporal Phosphoproteomic Data
Abstract
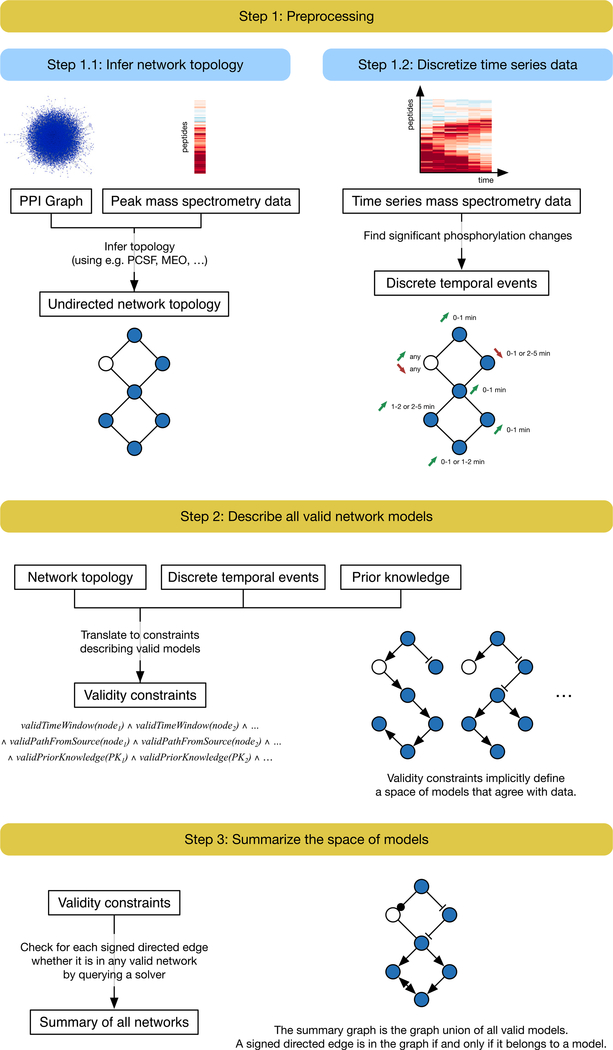
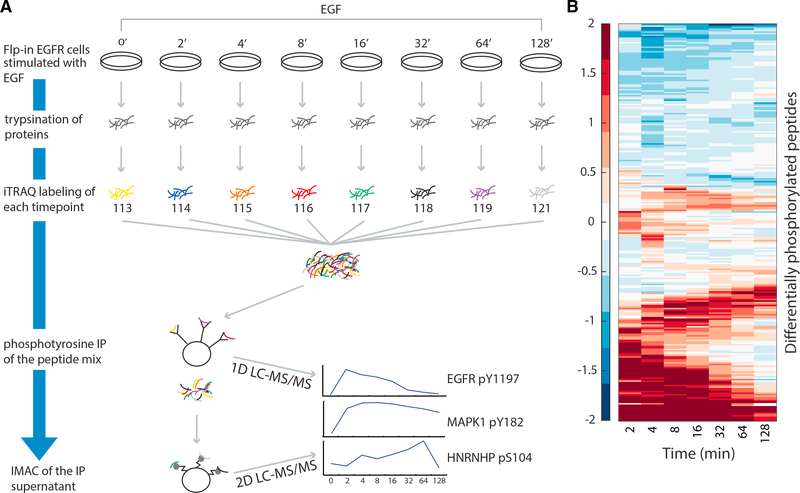
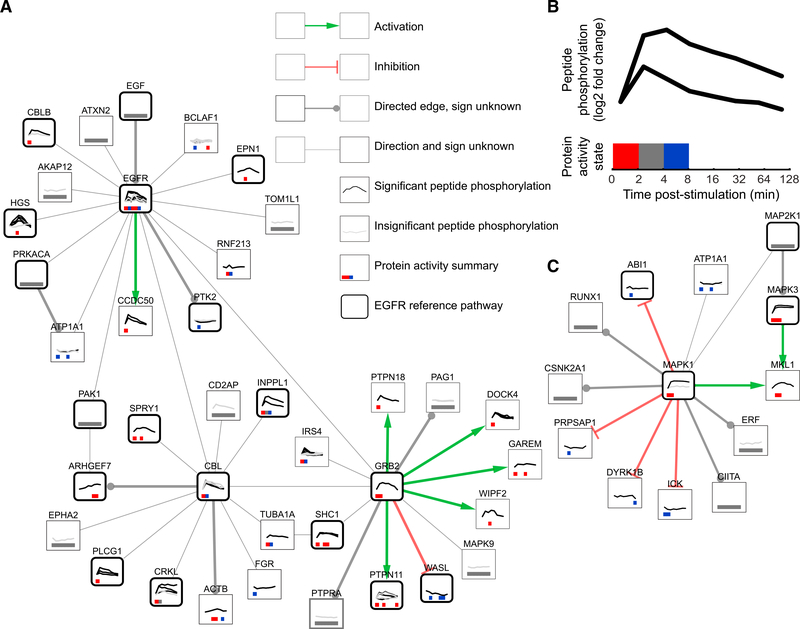
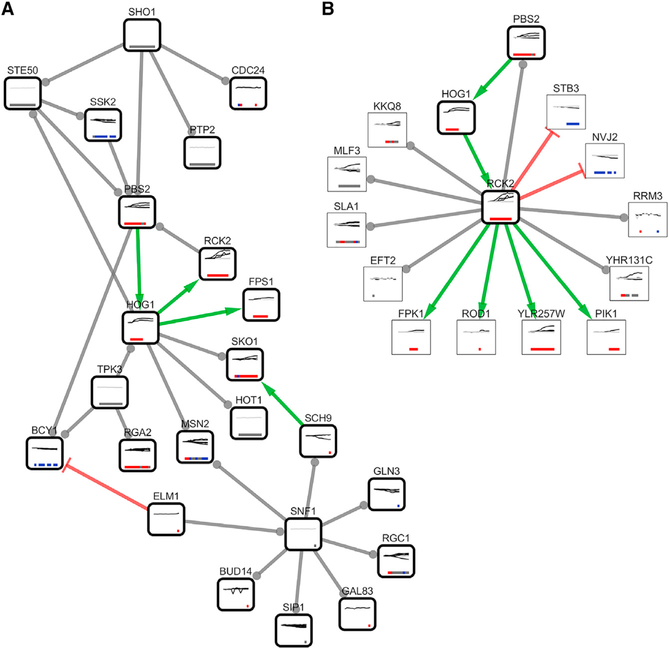
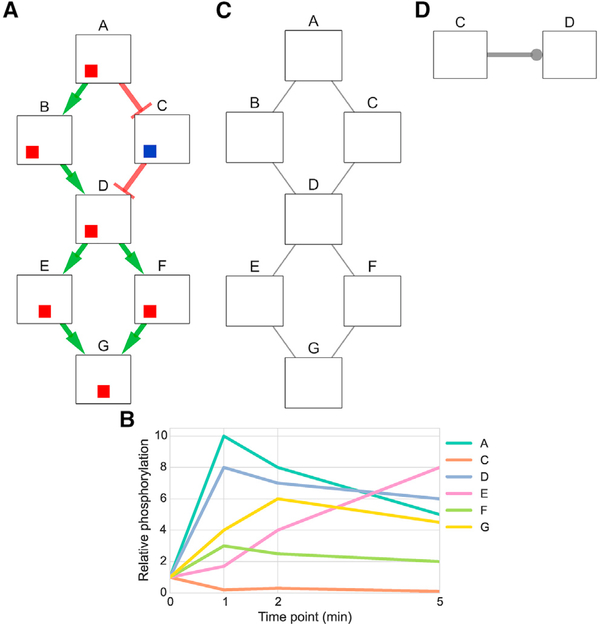
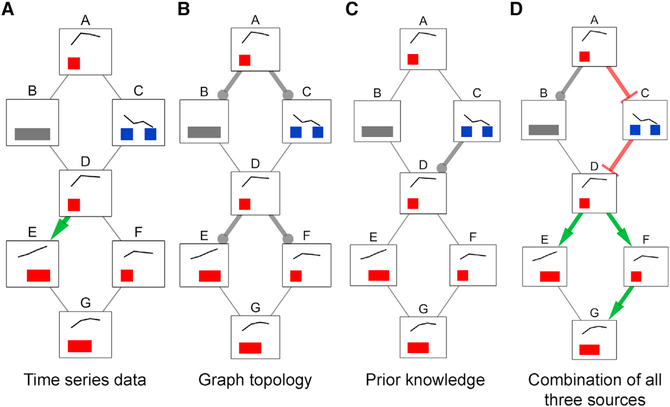
We present a method for automatically discovering signaling pathways from time-resolved phosphoproteomic data. The Temporal Pathway Synthesizer (TPS) algorithm uses constraint-solving techniques first developed in the context of formal verification to explore paths in an interaction network. It systematically eliminates all candidate structures for a signaling pathway where a protein is activated or inactivated before its upstream regulators. The algorithm can model more than one hundred thousand dynamic phosphosites and can discover pathway members that are not differentially phosphorylated. By analyzing temporal data, TPS defines signaling cascades without needing to experimentally perturb individual proteins. It recovers known pathways and proposes pathway connections when applied to the human epidermal growth factor and yeast osmotic stress responses. Independent kinase mutant studies validate predicted substrates in the TPS osmotic stress pathway.
Keywords: mass spectrometry; network algorithm; program synthesis; protein-protein interactions; time series phosphorylation.
Copyright © 2018 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
R.B. consults with NVIDIA.
Figures
References
-
- Bar-Joseph Z, Gitter A, and Simon I (2012). Studying and modelling dynamic biological processes using time-series gene expression data. Nat. Rev. Genet 13, 552–564. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
