

ATP hydrolysis-coupled peptide translocation mechanism of Mycobacterium tuberculosis ClpB
- PMID: 30257943
- PMCID: PMC6187150
- DOI: 10.1073/pnas.1810648115
ATP hydrolysis-coupled peptide translocation mechanism of Mycobacterium tuberculosis ClpB
Abstract
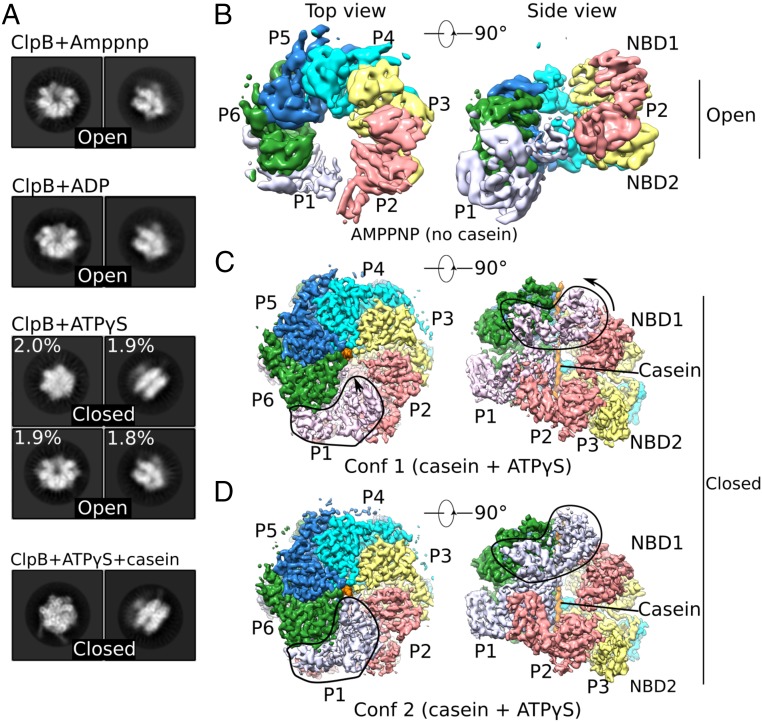
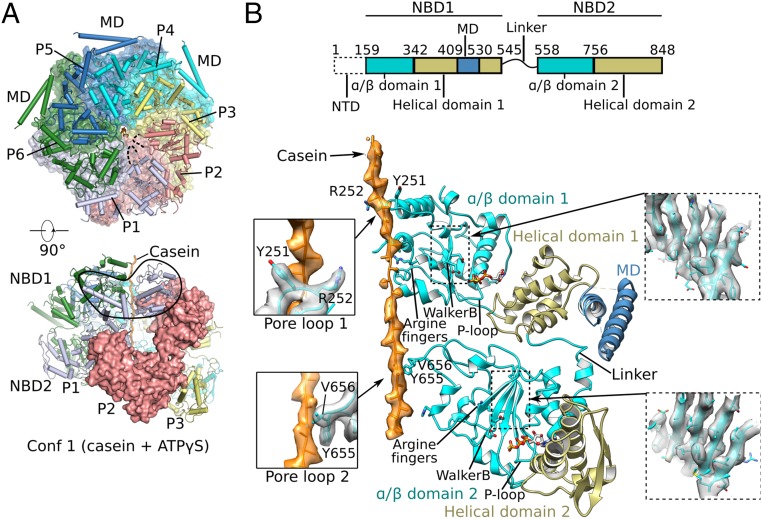
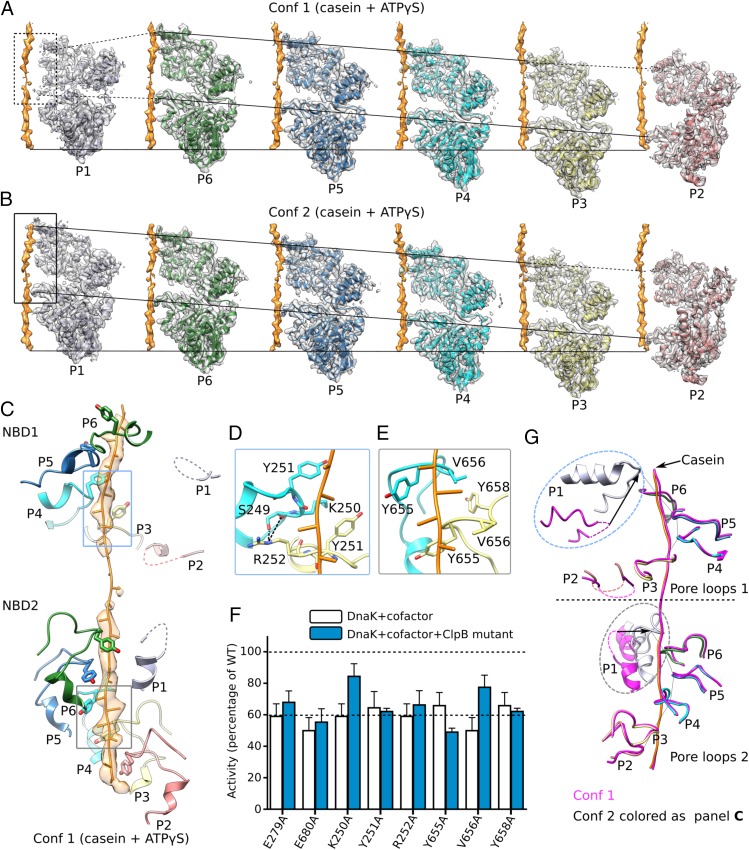
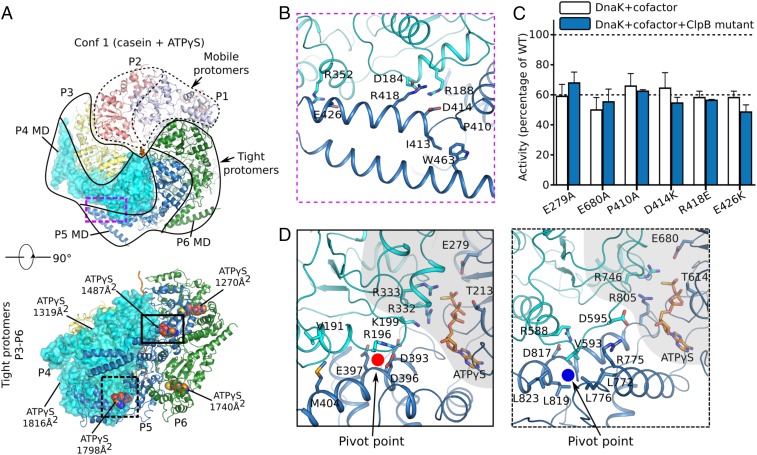
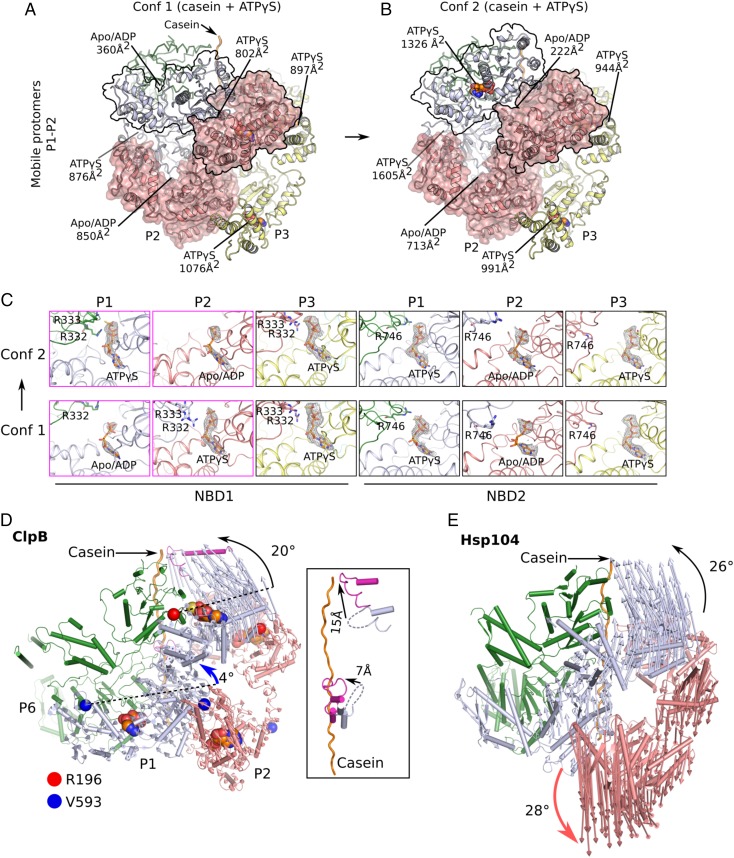
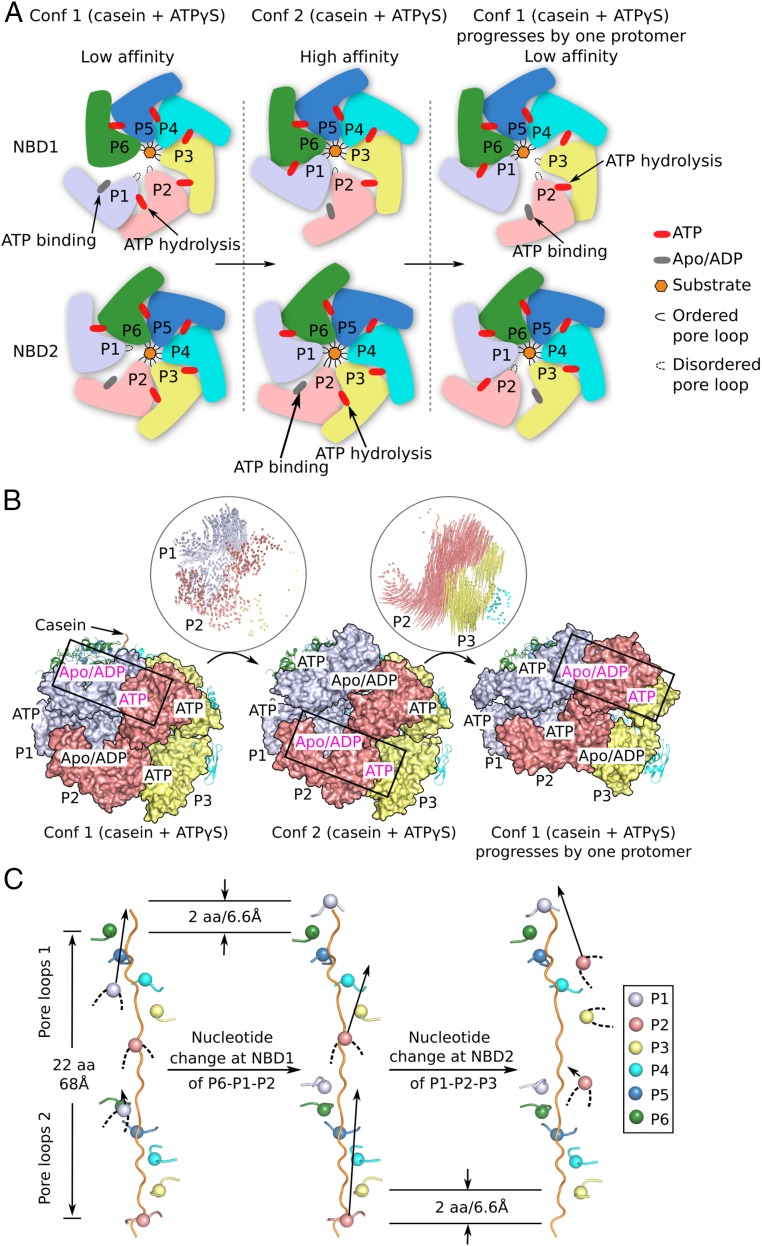
The protein disaggregase ClpB hexamer is conserved across evolution and has two AAA+-type nucleotide-binding domains, NBD1 and NBD2, in each protomer. In M. tuberculosis (Mtb), ClpB facilitates asymmetric distribution of protein aggregates during cell division to help the pathogen survive and persist within the host, but a mechanistic understanding has been lacking. Here we report cryo-EM structures at 3.8- to 3.9-Å resolution of Mtb ClpB bound to a model substrate, casein, in the presence of the weakly hydrolyzable ATP mimic adenosine 5'-[γ-thio]triphosphate. Mtb ClpB existed in solution in two closed-ring conformations, conformers 1 and 2. In both conformers, the 12 pore-loops on the 12 NTDs of the six protomers (P1-P6) were arranged similarly to a staircase around the bound peptide. Conformer 1 is a low-affinity state in which three of the 12 pore-loops (the protomer P1 NBD1 and NBD2 loops and the protomer P2 NBD1 loop) are not engaged with peptide. Conformer 2 is a high-affinity state because only one pore-loop (the protomer P2 NBD1 loop) is not engaged with the peptide. The resolution of the two conformations, along with their bound substrate peptides and nucleotides, enabled us to propose a nucleotide-driven peptide translocation mechanism of a bacterial ClpB that is largely consistent with several recent unfoldase structures, in particular with the eukaryotic Hsp104. However, whereas Hsp104's two NBDs move in opposing directions during one step of peptide translocation, in Mtb ClpB the two NBDs move only in the direction of translocation.
Keywords: AAA-ATPase; Mycobacterium tuberculosis; cryo-EM; disaggregase; proteostasis.
Copyright © 2018 the Author(s). Published by PNAS.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Structural basis for substrate gripping and translocation by the ClpB AAA+ disaggregase.Nat Commun. 2019 Jun 3;10(1):2393. doi: 10.1038/s41467-019-10150-y. Nat Commun. 2019. PMID: 31160557 Free PMC article.
-
Two-Step Activation Mechanism of the ClpB Disaggregase for Sequential Substrate Threading by the Main ATPase Motor.Cell Rep. 2019 Jun 18;27(12):3433-3446.e4. doi: 10.1016/j.celrep.2019.05.075. Cell Rep. 2019. PMID: 31216466 Free PMC article.
-
Structural basis for aggregate dissolution and refolding by the Mycobacterium tuberculosis ClpB-DnaK bi-chaperone system.Cell Rep. 2021 May 25;35(8):109166. doi: 10.1016/j.celrep.2021.109166. Cell Rep. 2021. PMID: 34038719 Free PMC article.
-
Stairway to translocation: AAA+ motor structures reveal the mechanisms of ATP-dependent substrate translocation.Protein Sci. 2020 Feb;29(2):407-419. doi: 10.1002/pro.3743. Epub 2019 Oct 17. Protein Sci. 2020. PMID: 31599052 Free PMC article. Review.
-
Fast dynamics shape the function of the AAA+ machine ClpB: lessons from single-molecule FRET spectroscopy.FEBS J. 2023 Jul;290(14):3496-3511. doi: 10.1111/febs.16539. Epub 2022 Jun 25. FEBS J. 2023. PMID: 35638578 Review.
Cited by
-
Conformational plasticity of the ClpAP AAA+ protease couples protein unfolding and proteolysis.Nat Struct Mol Biol. 2020 May;27(5):406-416. doi: 10.1038/s41594-020-0409-5. Epub 2020 Apr 20. Nat Struct Mol Biol. 2020. PMID: 32313240 Free PMC article.
-
The mycobacterial proteasomal ATPase Mpa forms a gapped ring to engage the 20S proteasome.J Biol Chem. 2021 Jan-Jun;296:100713. doi: 10.1016/j.jbc.2021.100713. Epub 2021 Apr 27. J Biol Chem. 2021. PMID: 33930464 Free PMC article.
-
Structural basis for distinct operational modes and protease activation in AAA+ protease Lon.Sci Adv. 2020 May 20;6(21):eaba8404. doi: 10.1126/sciadv.aba8404. eCollection 2020 May. Sci Adv. 2020. PMID: 32490208 Free PMC article.
-
Structures of the ATP-fueled ClpXP proteolytic machine bound to protein substrate.Elife. 2020 Feb 28;9:e52774. doi: 10.7554/eLife.52774. Elife. 2020. PMID: 32108573 Free PMC article.
-
Unmasking AlphaFold to integrate experiments and predictions in multimeric complexes.Nat Commun. 2024 Oct 9;15(1):8724. doi: 10.1038/s41467-024-52951-w. Nat Commun. 2024. PMID: 39379372 Free PMC article.
References
-
- Doyle SM, Genest O, Wickner S. Protein rescue from aggregates by powerful molecular chaperone machines. Nat Rev Mol Cell Biol. 2013;14:617–629. - PubMed
-
- Doyle SM, Wickner S. Hsp104 and ClpB: Protein disaggregating machines. Trends Biochem Sci. 2009;34:40–48. - PubMed
-
- Weibezahn J, et al. Thermotolerance requires refolding of aggregated proteins by substrate translocation through the central pore of ClpB. Cell. 2004;119:653–665. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
