

Genetic profile and onset features of 1005 patients with Charcot-Marie-Tooth disease in Japan
- PMID: 30257968
- PMCID: PMC6518473
- DOI: 10.1136/jnnp-2018-318839
Genetic profile and onset features of 1005 patients with Charcot-Marie-Tooth disease in Japan
Abstract
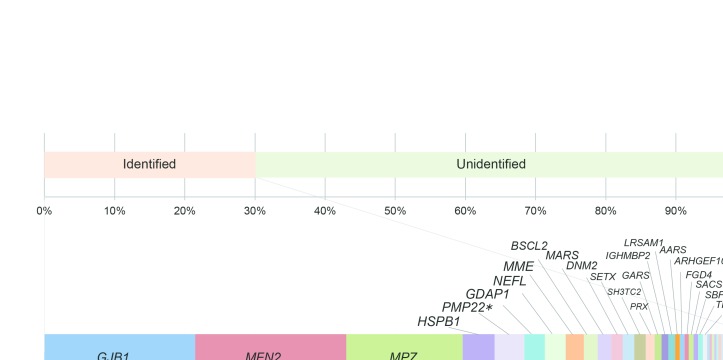
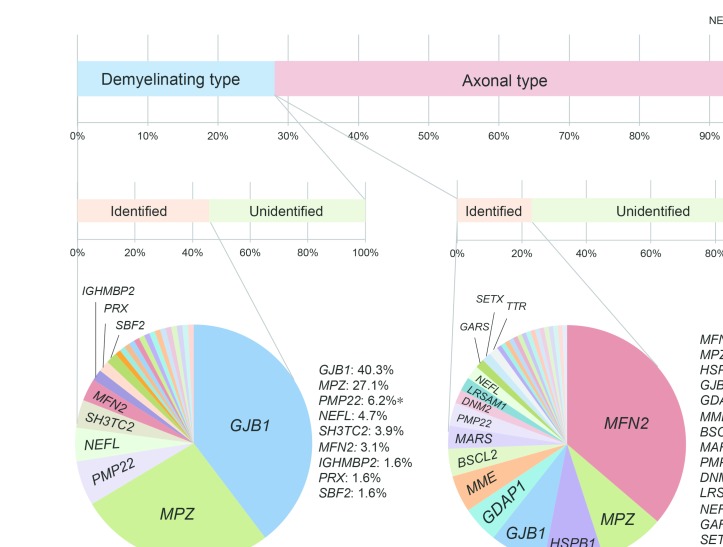
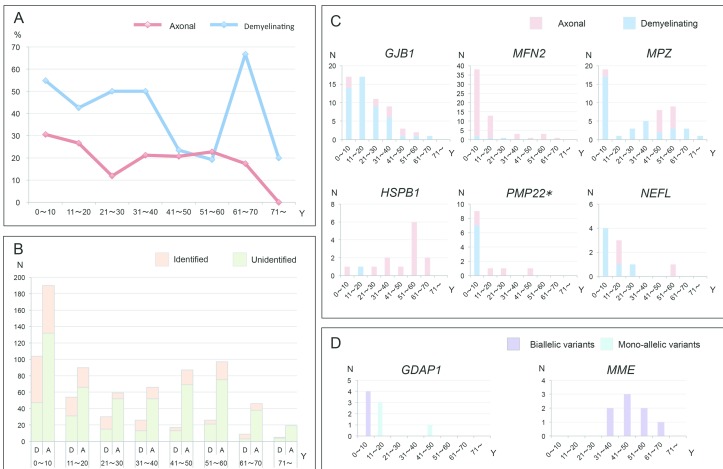
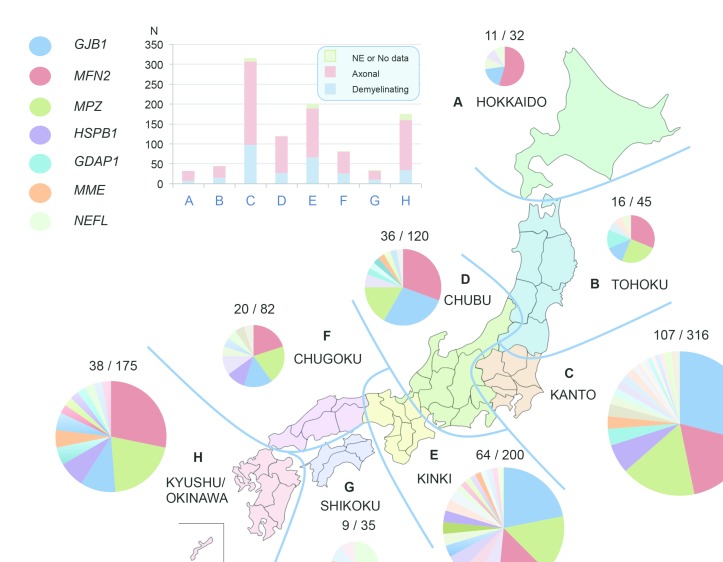
OBJECTIVE : To identify the genetic characteristics in a large-scale of patients with Charcot-Marie-Tooth disease (CMT). METHODS: From May 2012 to August 2016, we collected 1005 cases with suspected CMT throughout Japan, whereas PMP22 duplication/deletion were excluded in advance for demyelinating CMT cases. We performed next-generation sequencing targeting CMT-related gene panels using Illumina MiSeq or Ion Proton, then analysed the gene-specific onset age of the identified cases and geographical differences in terms of their genetic spectrum. RESULTS : From 40 genes, we identified pathogenic or likely pathogenic variants in 301 cases (30.0%). The most common causative genes were GJB1 (n=66, 21.9%), MFN2 (n=66, 21.9%) and MPZ (n=51, 16.9%). In demyelinating CMT, variants were detected in 45.7% cases, and the most common reasons were GJB1 (40.3%), MPZ (27.1%), PMP22 point mutations (6.2%) and NEFL (4.7%). Axonal CMT yielded a relatively lower detection rate (22.9%), and the leading causes, occupying 72.4%, were MFN2 (37.2%), MPZ (9.0%), HSPB1 (8.3%), GJB1 (7.7%), GDAP1 (5.1%) and MME (5.1%). First decade of life was found as the most common disease onset period, and early-onset CMT cases were most likely to receive a molecular diagnosis. Geographical distribution analysis indicated distinctive genetic spectrums in different regions of Japan. CONCLUSIONS : Our results updated the genetic profile within a large-scale of Japanese CMT cases. Subsequent analyses regarding onset age and geographical distribution advanced our understanding of CMT, which would be beneficial for clinicians.
Keywords: charcot-marie-tooth disease; gene panel; molecular epidemiology; next generation sequencing.
© Author(s) (or their employer(s)) 2019. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous