

Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome
- PMID: 30258076
- PMCID: PMC6158192
- DOI: 10.1038/s41598-018-32615-8
Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome
Erratum in
-
Author Correction: Direct RNA Sequencing of the Coding Complete Influenza A Virus Genome.Sci Rep. 2018 Oct 19;8(1):15746. doi: 10.1038/s41598-018-34067-6. Sci Rep. 2018. PMID: 30341398 Free PMC article.
Abstract
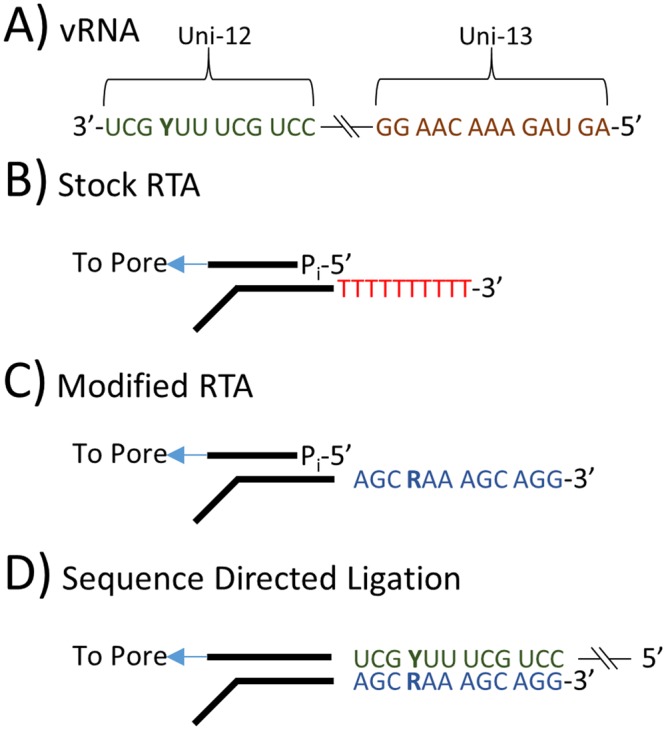
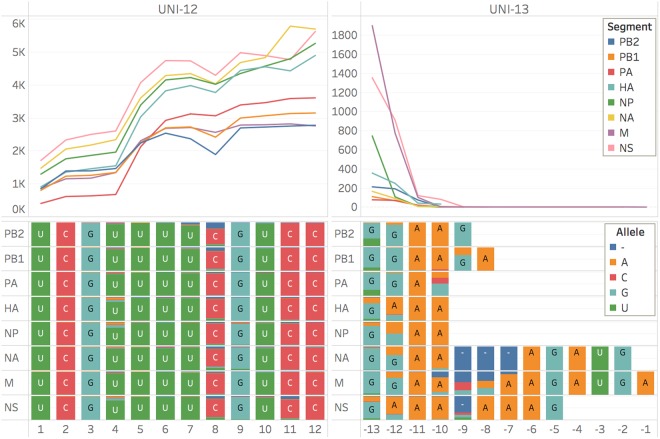
For the first time, a coding complete genome of an RNA virus has been sequenced in its original form. Previously, RNA was sequenced by the chemical degradation of radiolabeled RNA, a difficult method that produced only short sequences. Instead, RNA has usually been sequenced indirectly by copying it into cDNA, which is often amplified to dsDNA by PCR and subsequently analyzed using a variety of DNA sequencing methods. We designed an adapter to short highly conserved termini of the influenza A virus genome to target the (-) sense RNA into a protein nanopore on the Oxford Nanopore MinION sequencing platform. Utilizing this method with total RNA extracted from the allantoic fluid of influenza rA/Puerto Rico/8/1934 (H1N1) virus infected chicken eggs (EID50 6.8 × 109), we demonstrate successful sequencing of the coding complete influenza A virus genome with 100% nucleotide coverage, 99% consensus identity, and 99% of reads mapped to influenza A virus. By utilizing the same methodology one can redesign the adapter in order to expand the targets to include viral mRNA and (+) sense cRNA, which are essential to the viral life cycle, or other pathogens. This approach also has the potential to identify and quantify splice variants and base modifications, which are not practically measurable with current methods.
Conflict of interest statement
The authors declare no competing interests.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
