

Common genetic variants contribute to risk of rare severe neurodevelopmental disorders
- PMID: 30258228
- PMCID: PMC6726472
- DOI: 10.1038/s41586-018-0566-4
Common genetic variants contribute to risk of rare severe neurodevelopmental disorders
Abstract
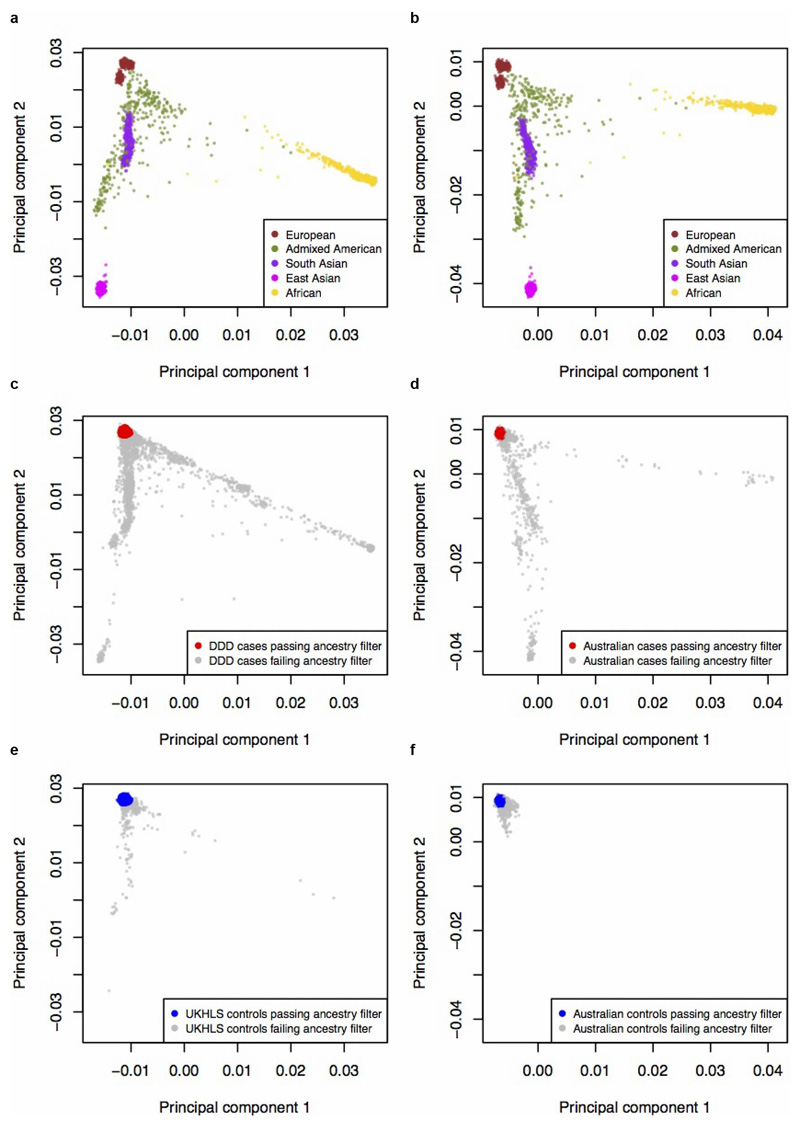
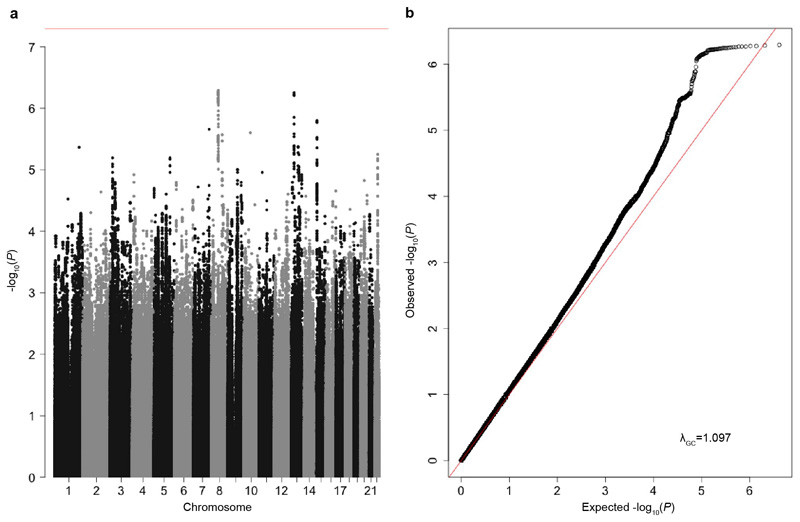
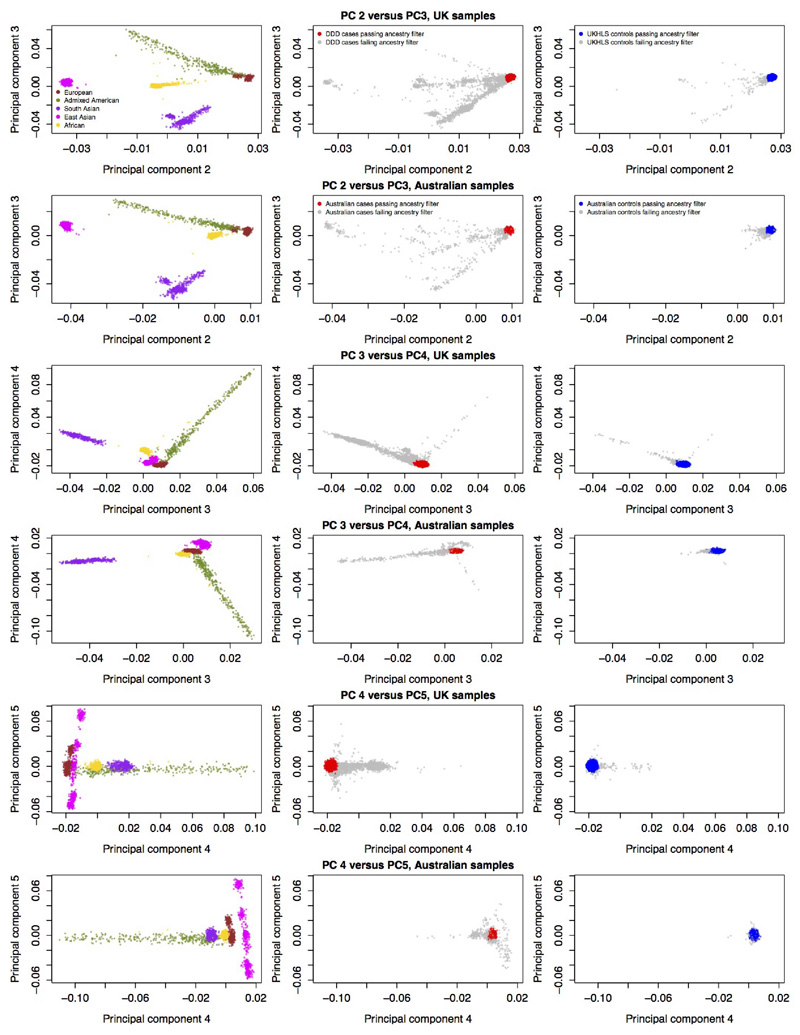
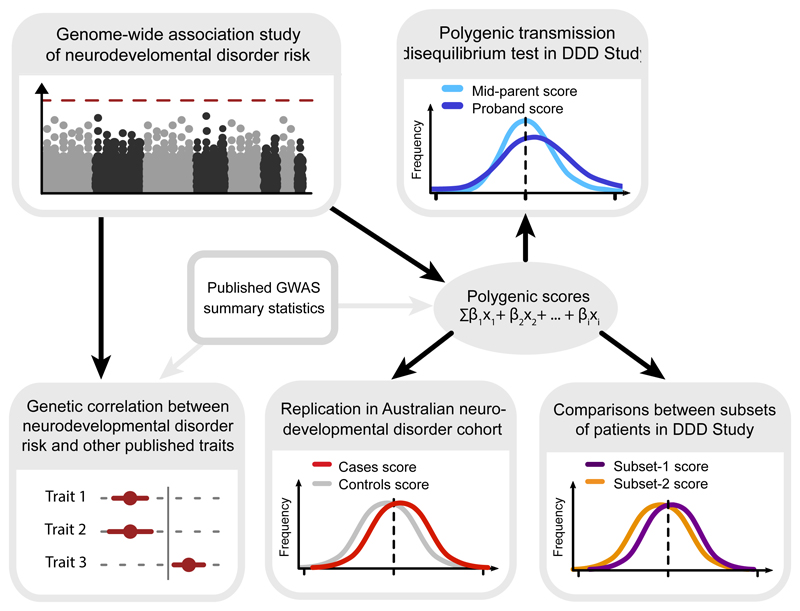
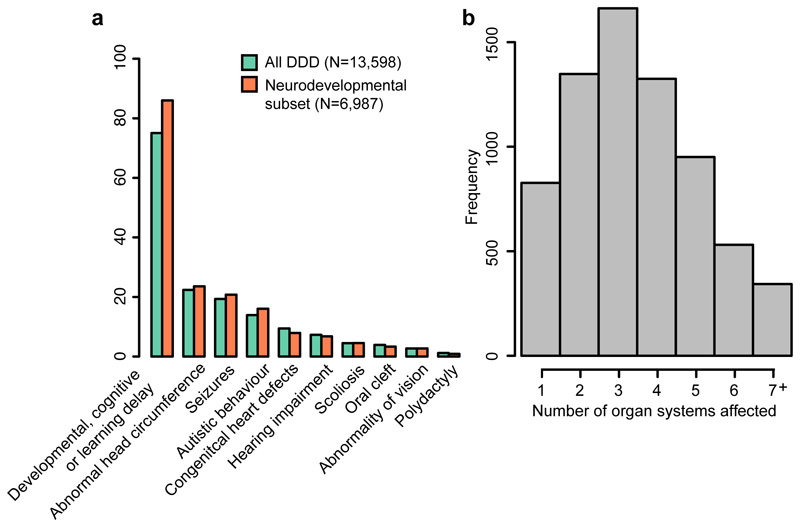
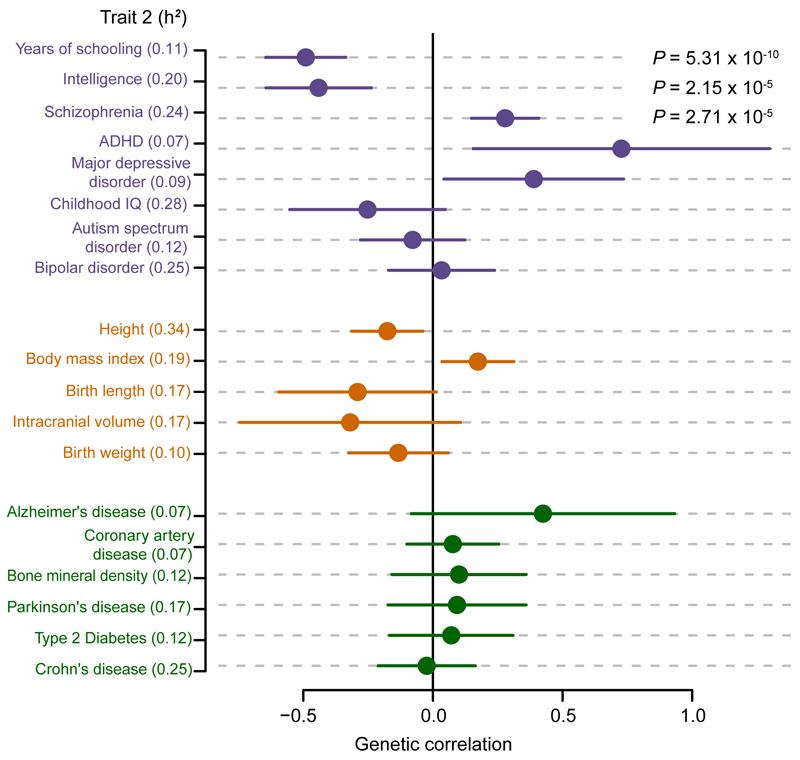
There are thousands of rare human disorders that are caused by single deleterious, protein-coding genetic variants1. However, patients with the same genetic defect can have different clinical presentations2-4, and some individuals who carry known disease-causing variants can appear unaffected5. Here, to understand what explains these differences, we study a cohort of 6,987 children assessed by clinical geneticists to have severe neurodevelopmental disorders such as global developmental delay and autism, often in combination with abnormalities of other organ systems. Although the genetic causes of these neurodevelopmental disorders are expected to be almost entirely monogenic, we show that 7.7% of variance in risk is attributable to inherited common genetic variation. We replicated this genome-wide common variant burden by showing, in an independent sample of 728 trios (comprising a child plus both parents) from the same cohort, that this burden is over-transmitted from parents to children with neurodevelopmental disorders. Our common-variant signal is significantly positively correlated with genetic predisposition to lower educational attainment, decreased intelligence and risk of schizophrenia. We found that common-variant risk was not significantly different between individuals with and without a known protein-coding diagnostic variant, which suggests that common-variant risk affects patients both with and without a monogenic diagnosis. In addition, previously published common-variant scores for autism, height, birth weight and intracranial volume were all correlated with these traits within our cohort, which suggests that phenotypic expression in individuals with monogenic disorders is affected by the same variants as in the general population. Our results demonstrate that common genetic variation affects both overall risk and clinical presentation in neurodevelopmental disorders that are typically considered to be monogenic.
Conflict of interest statement
M.E.H. is a co-founder of, consultant to, and holds shares in, Congenica Ltd, a genetics diagnostic company. J.C.B is an employee of Genomics plc.
Figures
References
-
- Balasubramanian M, et al. Delineating the phenotypic spectrum of Bainbridge-Ropers syndrome: 12 new patients with de novo, heterozygous, loss-of-function mutations inASXL3and review of published literature. J Med Genet. 2017;54:537–543. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
