

Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases
- PMID: 30258241
- PMCID: PMC6375686
- DOI: 10.1038/s41593-018-0238-6
Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases
Abstract
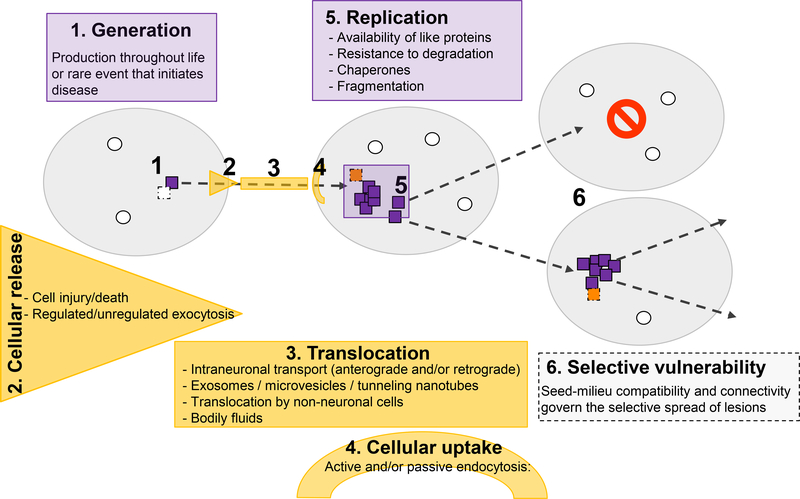
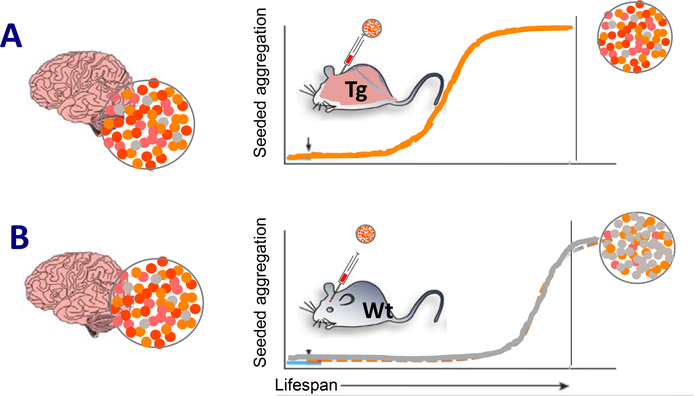
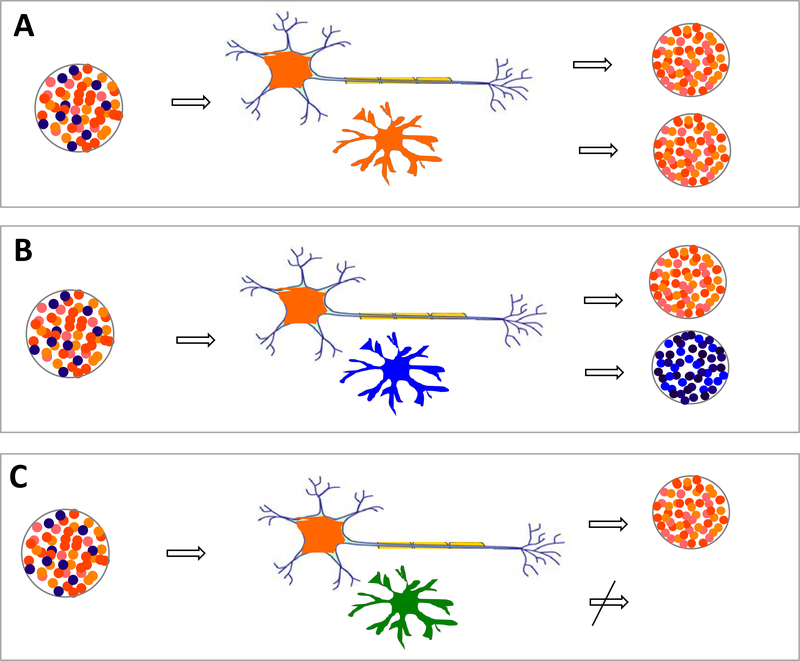
Many neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis, are characterized by the progressive appearance of abnormal proteinaceous assemblies in the nervous system. Studies in experimental systems indicate that the assemblies originate from the prion-like seeded aggregation of specific misfolded proteins that proliferate and amass to form the intracellular and/or extracellular lesions typical of each disorder. The host in which the proteopathic seeds arise provides the biochemical and physiological environment that either supports or restricts their emergence, proliferation, self-assembly, and spread. Multiple mechanisms influence the spatiotemporal spread of seeds and the nature of the resulting lesions, one of which is the cellular uptake, release, and transport of seeds along neural pathways and networks. The characteristics of cells and regions in the affected network govern their vulnerability and thereby influence the neuropathological and clinical attributes of the disease. The propagation of pathogenic protein assemblies within the nervous system is thus determined by the interaction of the proteopathic agent and the host milieu.
Conflict of interest statement
Figures
References
-
- Paget S The distribution of secondary growths in cancer of the breast. The Lancet 133, 571–573 (1889). - PubMed
-
- Prusiner SB Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144 (1982). - PubMed
-
- Mead S & Reilly MM A new prion disease: relationship with central and peripheral amyloidoses. Nat. Rev. Neurol 11, 90–97 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
