

SEG - A Software Program for Finding Somatic Copy Number Alterations in Whole Genome Sequencing Data of Cancer
- PMID: 30258547
- PMCID: PMC6154469
- DOI: 10.1016/j.csbj.2018.09.001
SEG - A Software Program for Finding Somatic Copy Number Alterations in Whole Genome Sequencing Data of Cancer
Abstract
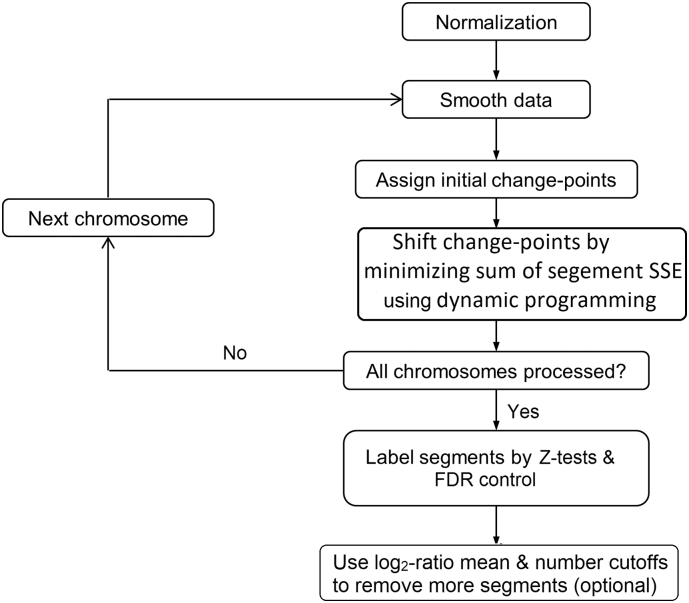
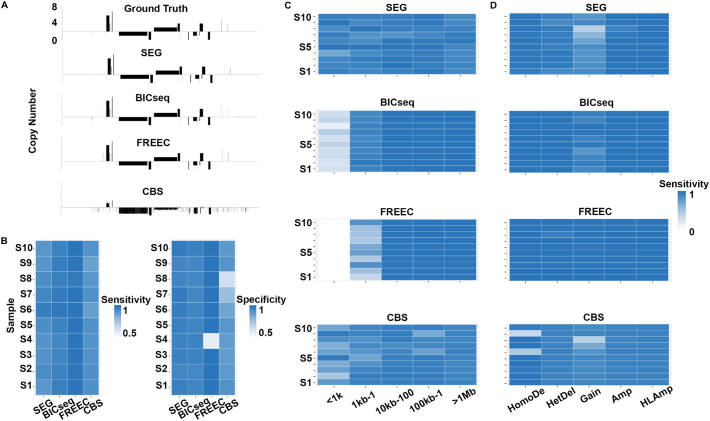
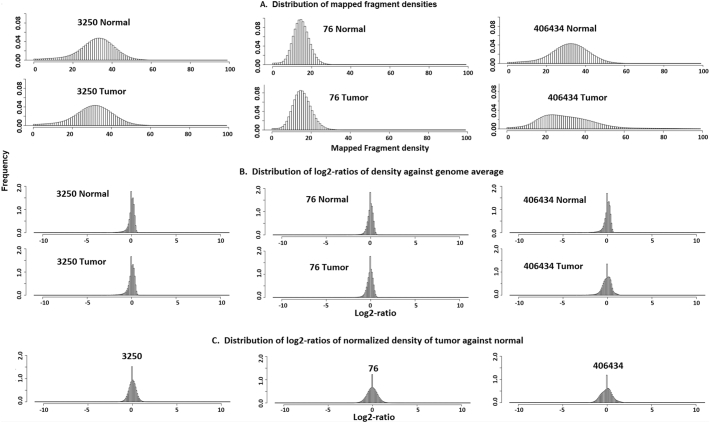
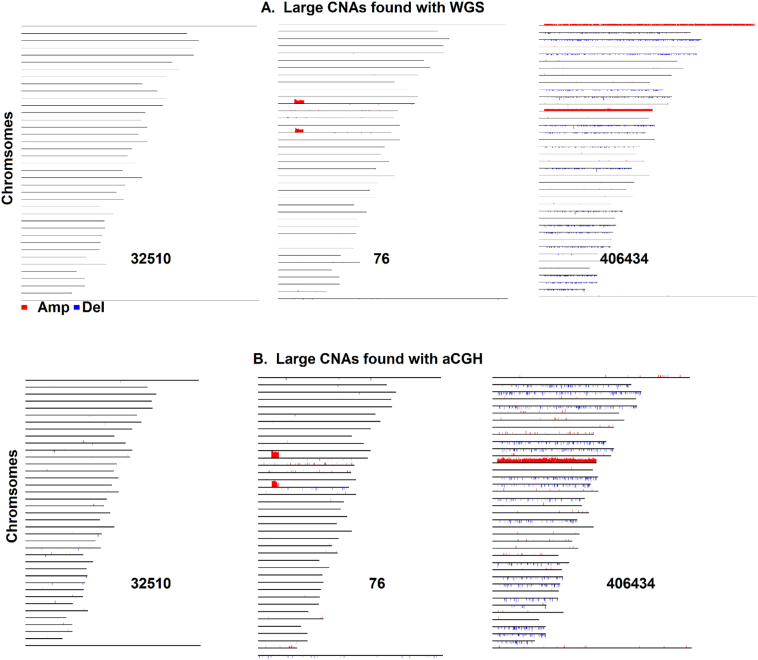
As next-generation sequencing technology advances and the cost decreases, whole genome sequencing (WGS) has become the preferred platform for the identification of somatic copy number alteration (CNA) events in cancer genomes. To more effectively decipher these massive sequencing data, we developed a software program named SEG, shortened from the word "segment". SEG utilizes mapped read or fragment density for CNA discovery. To reduce CNA artifacts arisen from sequencing and mapping biases, SEG first normalizes the data by taking the log2-ratio of each tumor density against its matching normal density. SEG then uses dynamic programming to find change-points among a contiguous log2-ratio data series along a chromosome, dividing the chromosome into different segments. SEG finally identifies those segments having CNA. Our analyses with both simulated and real sequencing data indicate that SEG finds more small CNAs than other published software tools.
Keywords: Cancer; SEG; Somatic Copy Number Alteration; Whole Genome Sequencing.
Figures
Similar articles
-
Hierarchical discovery of large-scale and focal copy number alterations in low-coverage cancer genomes.BMC Bioinformatics. 2020 Apr 16;21(1):147. doi: 10.1186/s12859-020-3480-3. BMC Bioinformatics. 2020. PMID: 32299346 Free PMC article.
-
Copy number alterations detected by whole-exome and whole-genome sequencing of esophageal adenocarcinoma.Hum Genomics. 2015 Sep 15;9(1):22. doi: 10.1186/s40246-015-0044-0. Hum Genomics. 2015. PMID: 26374103 Free PMC article.
-
CLImAT-HET: detecting subclonal copy number alterations and loss of heterozygosity in heterogeneous tumor samples from whole-genome sequencing data.BMC Med Genomics. 2017 Mar 15;10(1):15. doi: 10.1186/s12920-017-0255-4. BMC Med Genomics. 2017. PMID: 28298214 Free PMC article.
-
Using SAAS-CNV to Detect and Characterize Somatic Copy Number Alterations in Cancer Genomes from Next Generation Sequencing and SNP Array Data.Methods Mol Biol. 2018;1833:29-47. doi: 10.1007/978-1-4939-8666-8_2. Methods Mol Biol. 2018. PMID: 30039361 Review.
-
Cancer whole-genome sequencing: present and future.Oncogene. 2015 Dec 3;34(49):5943-50. doi: 10.1038/onc.2015.90. Epub 2015 Mar 30. Oncogene. 2015. PMID: 25823020 Review.
Cited by
-
Identification of Copy Number Alterations from Next-Generation Sequencing Data.Adv Exp Med Biol. 2022;1361:55-74. doi: 10.1007/978-3-030-91836-1_4. Adv Exp Med Biol. 2022. PMID: 35230683
-
A Kmer-based paired-end read de novo assembler and genotyper for canine MHC class I genotyping.iScience. 2023 Jan 16;26(2):105996. doi: 10.1016/j.isci.2023.105996. eCollection 2023 Feb 17. iScience. 2023. PMID: 36798440 Free PMC article.
-
Leading the pack: Best practices in comparative canine cancer genomics to inform human oncology.Vet Comp Oncol. 2023 Dec;21(4):565-577. doi: 10.1111/vco.12935. Epub 2023 Oct 1. Vet Comp Oncol. 2023. PMID: 37778398 Free PMC article. Review.
-
Canine tumor mutational burden is correlated with TP53 mutation across tumor types and breeds.Nat Commun. 2021 Aug 3;12(1):4670. doi: 10.1038/s41467-021-24836-9. Nat Commun. 2021. PMID: 34344882 Free PMC article.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials