

Personalized Tumor RNA Loaded Lipid-Nanoparticles Prime the Systemic and Intratumoral Milieu for Response to Cancer Immunotherapy
- PMID: 30259750
- PMCID: PMC6597257
- DOI: 10.1021/acs.nanolett.8b02179
Personalized Tumor RNA Loaded Lipid-Nanoparticles Prime the Systemic and Intratumoral Milieu for Response to Cancer Immunotherapy
Abstract
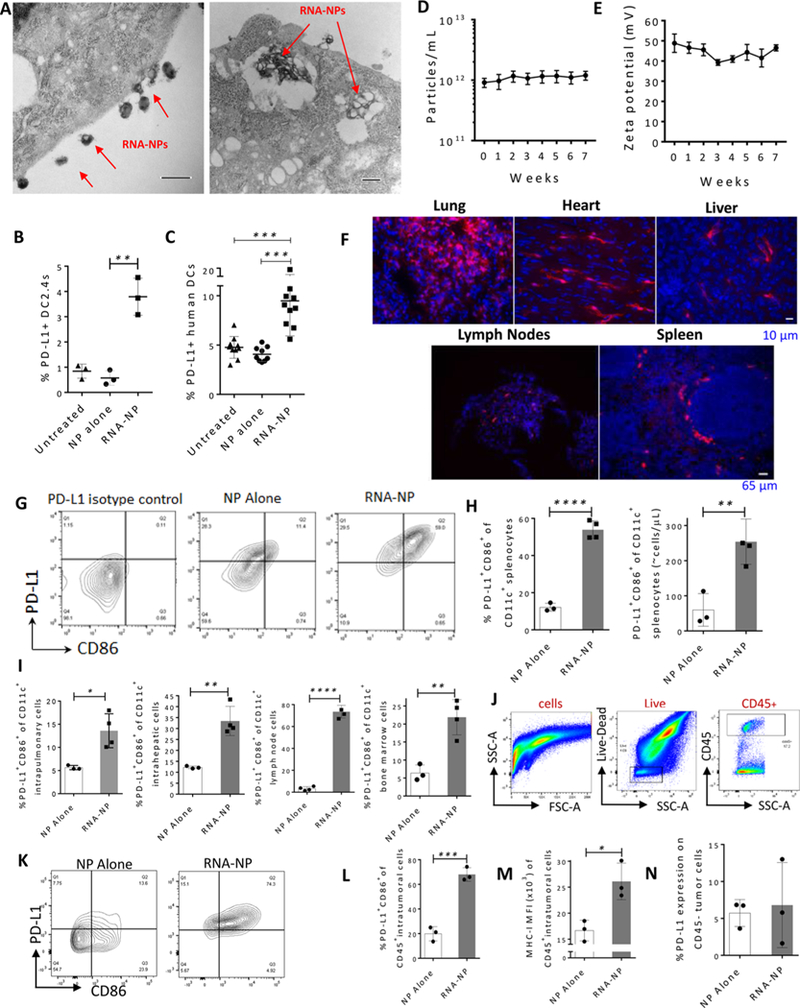
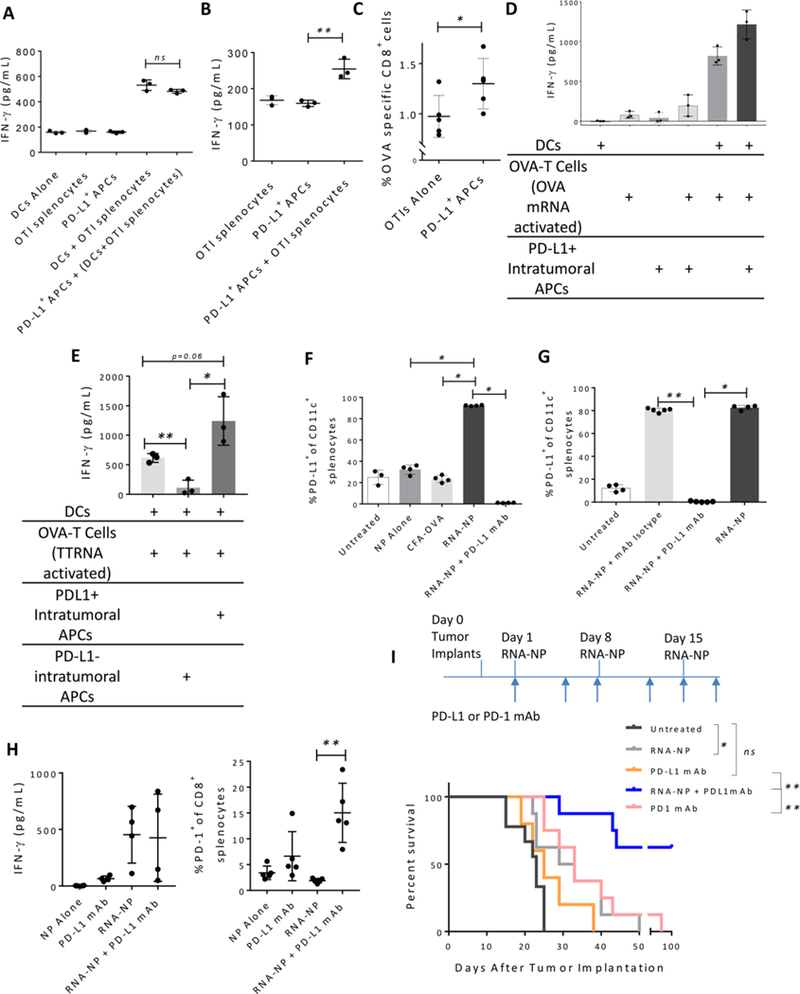
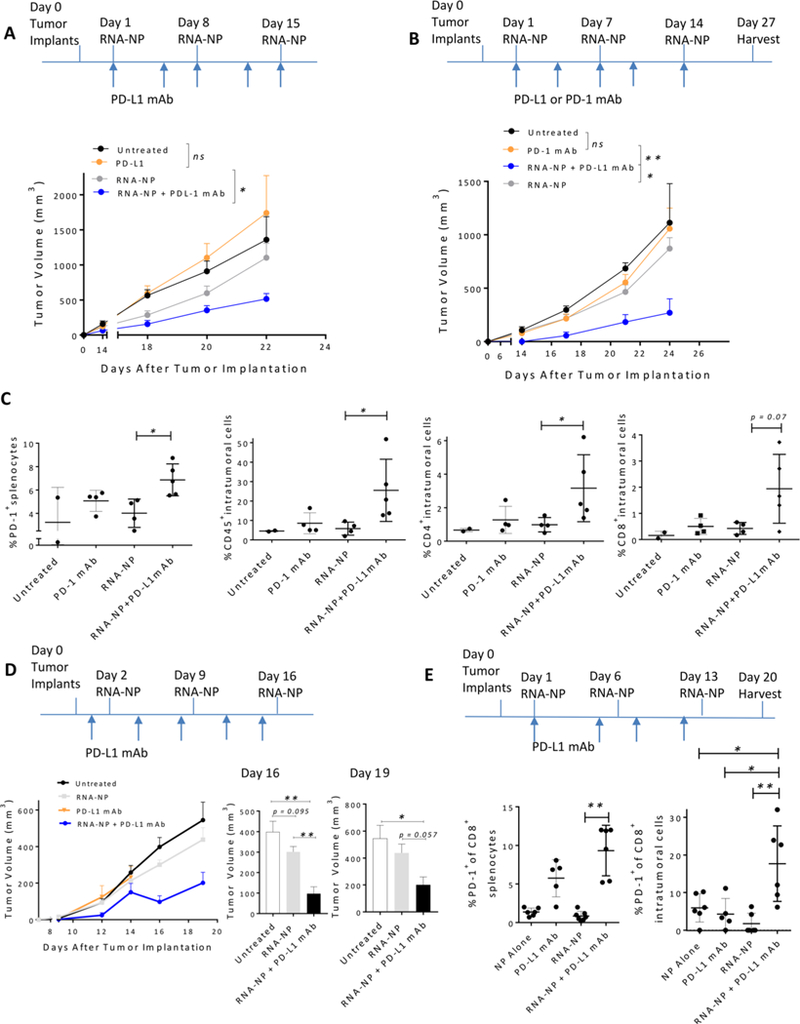
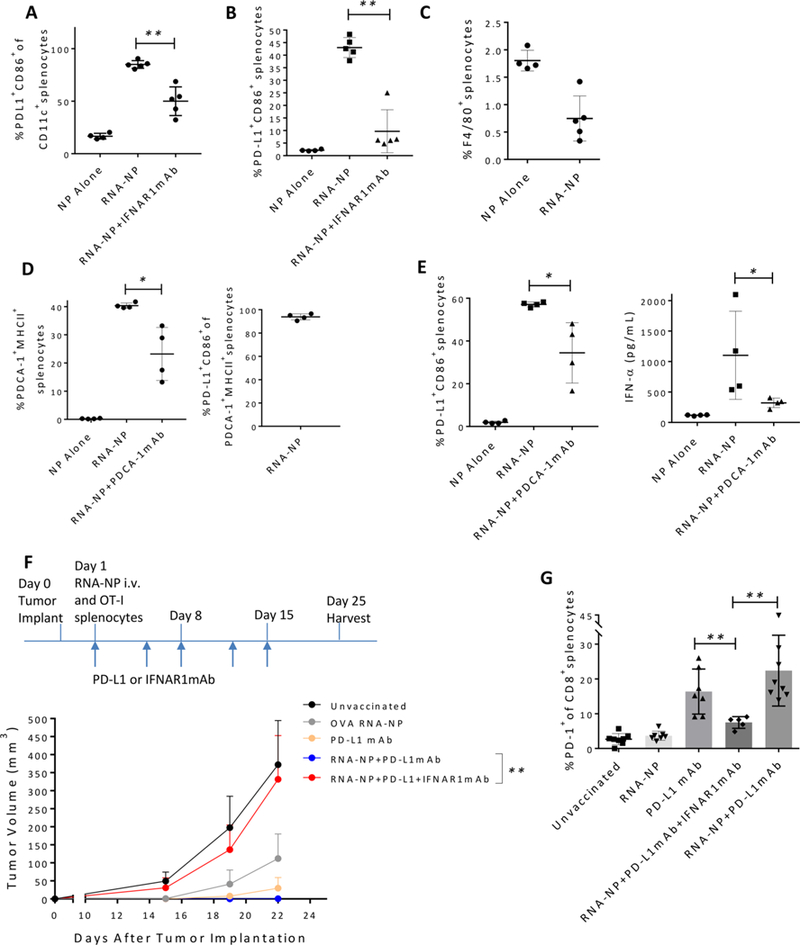
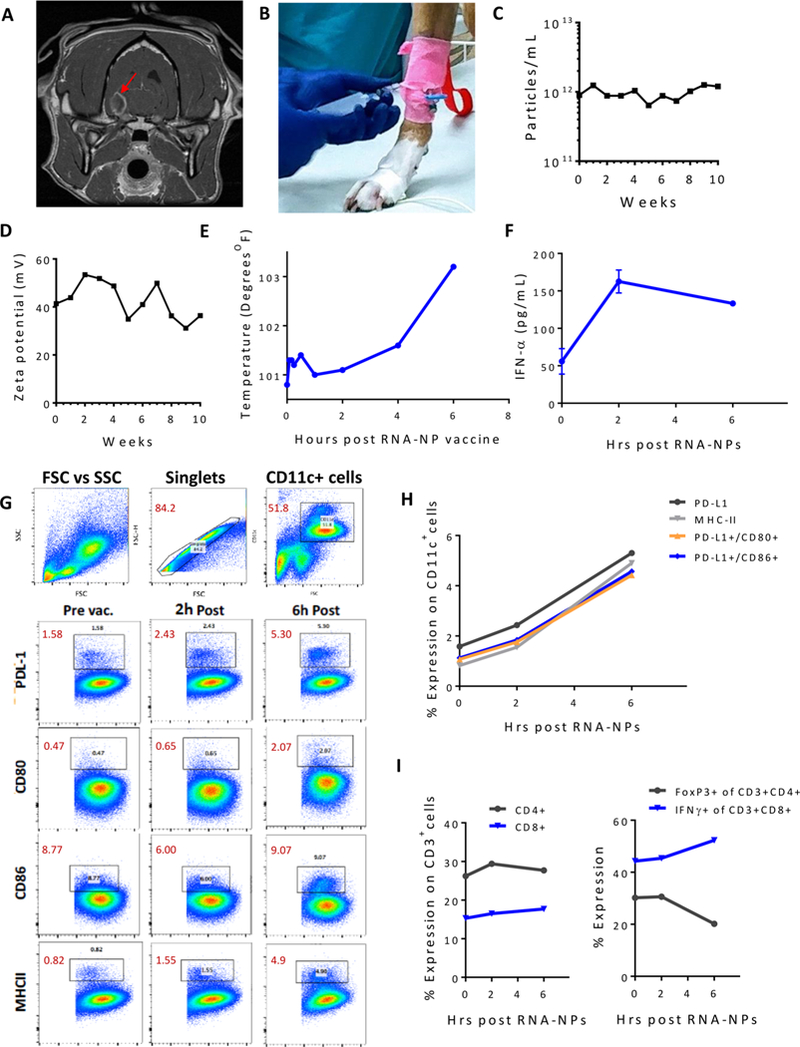
Translation of nanoparticles (NPs) into human clinical trials for patients with refractory cancers has lagged due to unknown biologic reactivities of novel NP designs. To overcome these limitations, simple well-characterized mRNA lipid-NPs have been developed as cancer immunotherapeutic vaccines. While the preponderance of RNA lipid-NPs encoding for tumor-associated antigens or neoepitopes have been designed to target lymphoid organs, they remain encumbered by the profound intratumoral and systemic immunosuppression that may stymie an activated T cell response. Herein, we show that systemic localization of untargeted tumor RNA (derived from whole transcriptome) encapsulated in lipid-NPs, with excess positive charge, primes the peripheral and intratumoral milieu for response to immunotherapy. In immunologically resistant tumor models, these RNA-NPs activate the preponderance of systemic and intratumoral myeloid cells (characterized by coexpression of PD-L1 and CD86). Addition of immune checkpoint inhibitors (ICIs) (to animals primed with RNA-NPs) augments peripheral/intratumoral PD-1+CD8+ cells and mediates synergistic antitumor efficacy in settings where ICIs alone do not confer therapeutic benefit. These synergistic effects are mediated by type I interferon released from plasmacytoid dendritic cells (pDCs). In translational studies, personalized mRNA-NPs were safe and active in a client-owned canine with a spontaneous malignant glioma. In summary, we demonstrate widespread immune activation from tumor loaded RNA-NPs concomitant with inducible PD-L1 expression that can be therapeutically exploited. While immunotherapy remains effective for only a subset of cancer patients, combination therapy with systemic immunomodulating RNA-NPs may broaden its therapeutic potency.
Keywords: RNA nanoparticles; cancer immunotherapy; cancer vaccines; immune checkpoint inhibitors; liposomes; personalized therapy.
Figures
References
-
- Brahmer JR; Tykodi SS; Chow LQ; Hwu WJ; Topalian SL; Hwu P; Drake CG; Camacho LH; Kauh J; Odunsi K; Pitot HC; Hamid O; Bhatia S; Martins R; Eaton K; Chen S; Salay TM; Alaparthy S; Grosso JF; Korman AJ; Parker SM; Agrawal S; Goldberg SM; Pardoll DM; Gupta A; Wigginton JM N. Engl. J. Med. 2012, 366 (26), 2455–65. - PMC - PubMed
-
- Borghaei H; Paz-Ares L; Horn L; Spigel DR; Steins M; Ready NE; Chow LQ; Vokes EE; Felip E; Holgado E; Barlesi F; Kohlhaufl M; Arrieta O; Burgio MA; Fayette J; Lena H; Poddubskaya E; Gerber DE; Gettinger SN; Rudin CM; Rizvi N; Crino L; Blumenschein GR Jr.; Antonia SJ; Dorange C; Harbison CT; Graf Finckenstein F; Brahmer JR N. Engl. J. Med. 2015, 373 (17), 1627–39. - PMC - PubMed
-
- Topalian SL; Hodi FS; Brahmer JR; Gettinger SN; Smith DC; McDermott DF; Powderly JD; Carvajal RD; Sosman JA; Atkins MB; Leming PD; Spigel DR; Antonia SJ; Horn L; Drake CG; Pardoll DM; Chen L; Sharfman WH; Anders RA; Taube JM; McMiller TL; Xu H; Korman AJ; Jure-Kunkel M; Agrawal S; McDonald D; Kollia GD; Gupta A; Wigginton JM; Sznol M N. Engl. J. Med. 2012, 366 (26), 2443–54. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
