

PLK1 stabilizes a MYC-dependent kinase network in aggressive B cell lymphomas
- PMID: 30260324
- PMCID: PMC6264635
- DOI: 10.1172/JCI122533
PLK1 stabilizes a MYC-dependent kinase network in aggressive B cell lymphomas
Abstract
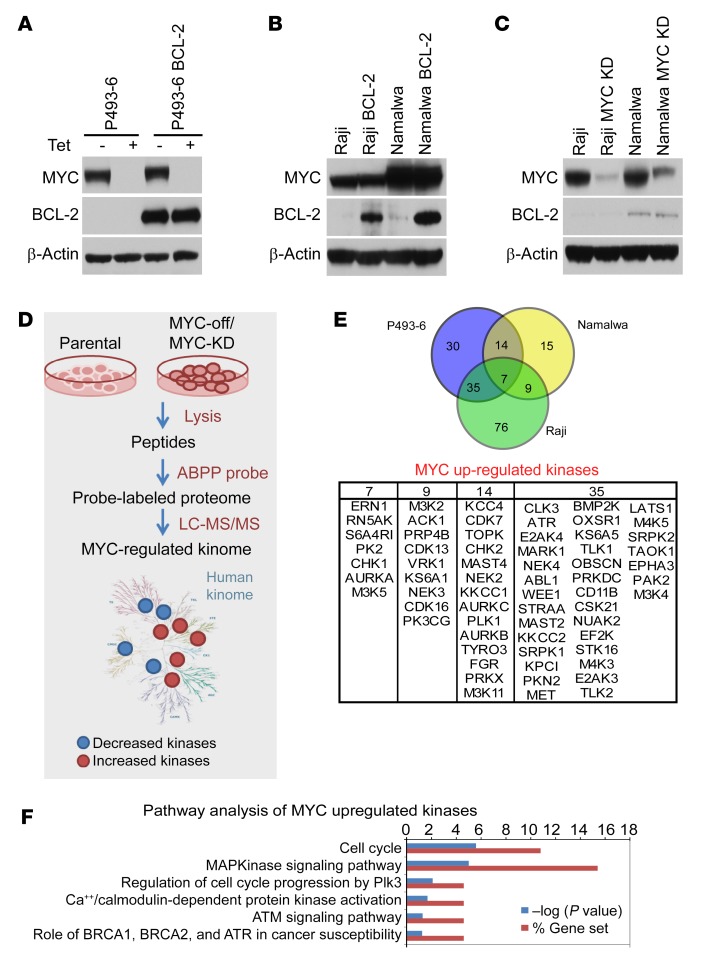
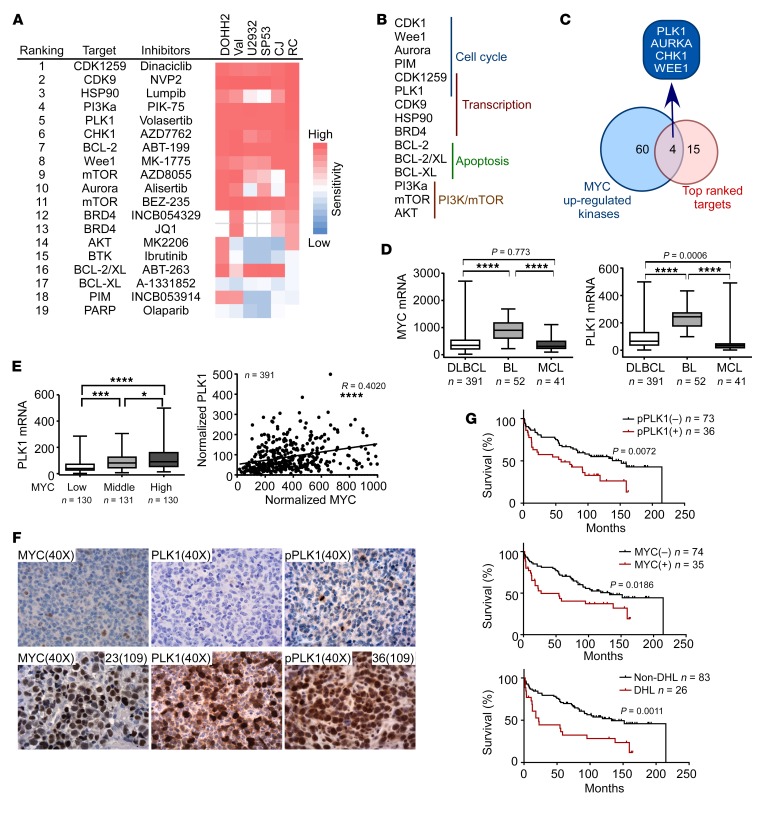
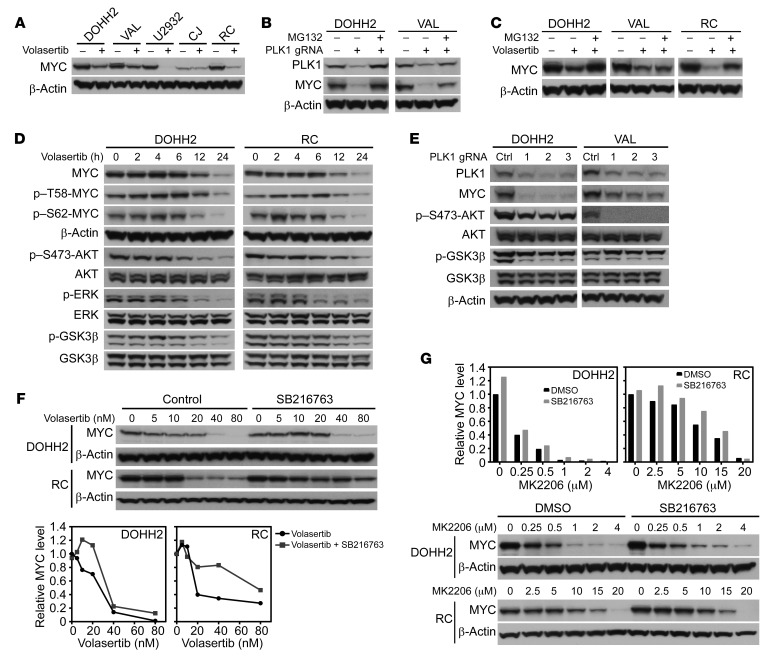
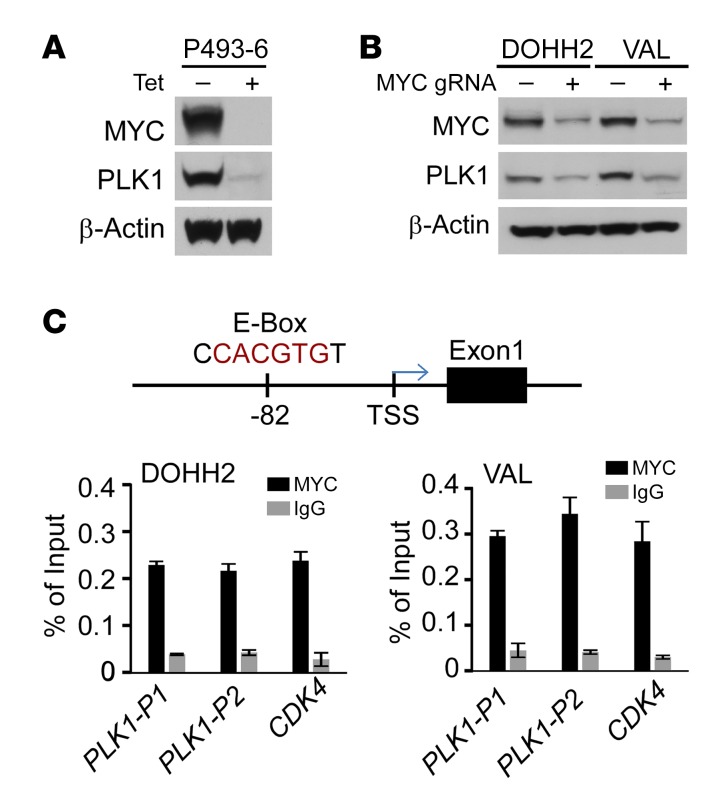
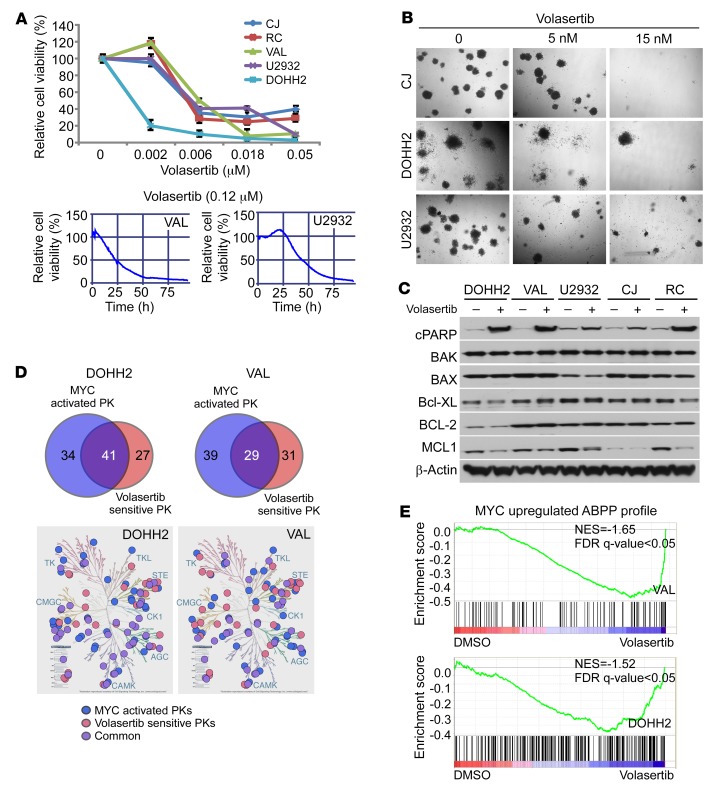
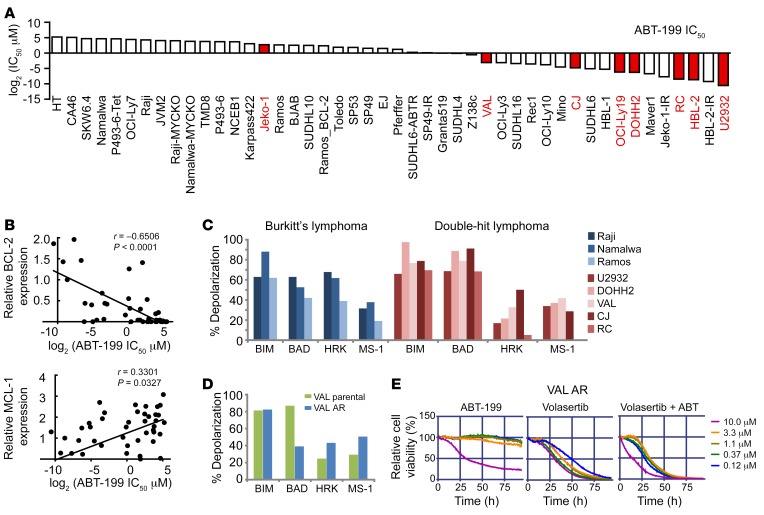
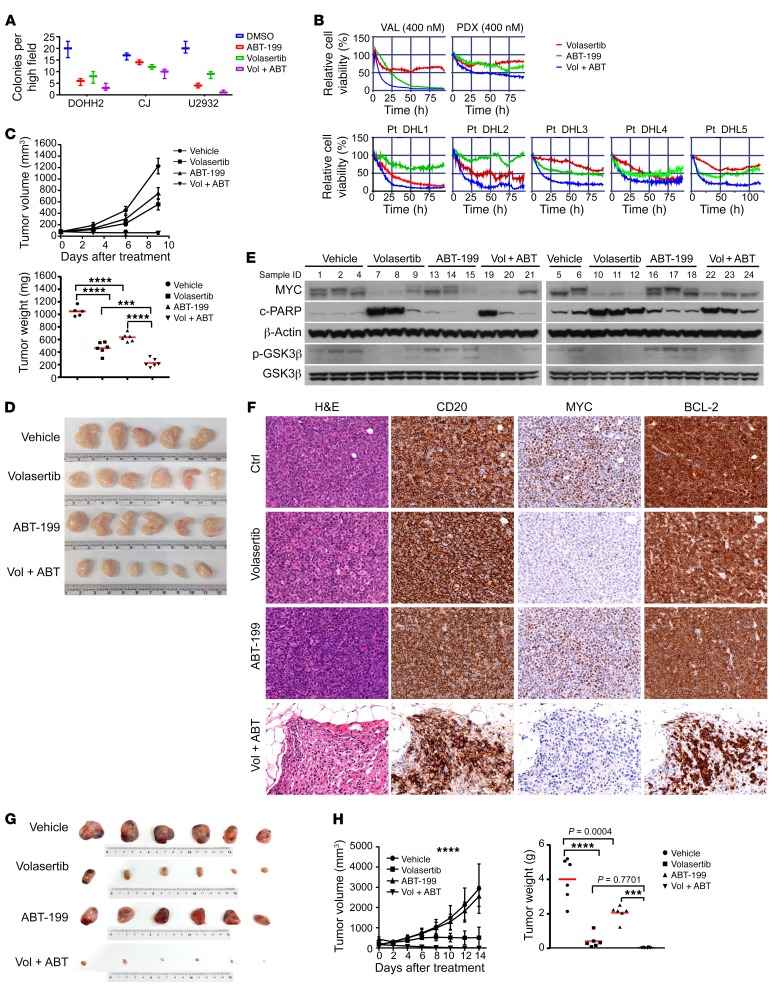
Concordant activation of MYC and BCL-2 oncoproteins in double-hit lymphoma (DHL) results in aggressive disease that is refractory to treatment. By integrating activity-based proteomic profiling and drug screens, polo-like kinase-1 (PLK1) was identified as an essential regulator of the MYC-dependent kinome in DHL. Notably, PLK1 was expressed at high levels in DHL, correlated with MYC expression, and connoted poor outcome. Further, PLK1 signaling augmented MYC protein stability, and in turn, MYC directly induced PLK1 transcription, establishing a feed-forward MYC-PLK1 circuit in DHL. Finally, inhibition of PLK1 triggered degradation of MYC and of the antiapoptotic protein MCL-1, and PLK1 inhibitors showed synergy with BCL-2 antagonists in blocking DHL cell growth, survival, and tumorigenicity, supporting clinical targeting of PLK1 in DHL.
Keywords: Cancer; Hematology; Lymphomas; Molecular biology; Oncology.
Conflict of interest statement
Figures
Comment in
-
PLK1: a promising and previously unexplored target in double-hit lymphoma.J Clin Invest. 2018 Dec 3;128(12):5206-5208. doi: 10.1172/JCI124919. Epub 2018 Nov 5. J Clin Invest. 2018. PMID: 30395539 Free PMC article.
Similar articles
-
PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy.Cancer Discov. 2013 Oct;3(10):1156-71. doi: 10.1158/2159-8290.CD-12-0595. Epub 2013 Jul 25. Cancer Discov. 2013. PMID: 23887393
-
The contribution of MYC and PLK1 expression to proliferative capacity in diffuse large B-cell lymphoma.Leuk Lymphoma. 2019 Dec;60(13):3214-3224. doi: 10.1080/10428194.2019.1633629. Epub 2019 Jul 1. Leuk Lymphoma. 2019. PMID: 31259656
-
Establishment and characterization of a novel MYC/BCL2 "double-hit" diffuse large B cell lymphoma cell line, RC.J Hematol Oncol. 2015 Oct 29;8:121. doi: 10.1186/s13045-015-0218-1. J Hematol Oncol. 2015. PMID: 26515759 Free PMC article.
-
The role of aurora A and polo-like kinases in high-risk lymphomas.Blood Adv. 2019 Jun 11;3(11):1778-1787. doi: 10.1182/bloodadvances.2019000232. Blood Adv. 2019. PMID: 31186254 Free PMC article. Review.
-
MYC-associated and double-hit lymphomas: a review of pathobiology, prognosis, and therapeutic approaches.Cancer. 2014 Dec 15;120(24):3884-95. doi: 10.1002/cncr.28899. Epub 2014 Jul 24. Cancer. 2014. PMID: 25060588 Review.
Cited by
-
The complexity of p53-mediated metabolic regulation in tumor suppression.Semin Cancer Biol. 2022 Oct;85:4-32. doi: 10.1016/j.semcancer.2021.03.010. Epub 2021 Mar 27. Semin Cancer Biol. 2022. PMID: 33785447 Free PMC article. Review.
-
Targeting ST8SIA6-AS1 counteracts KRASG12C inhibitor resistance through abolishing the reciprocal activation of PLK1/c-Myc signaling.Exp Hematol Oncol. 2023 Dec 16;12(1):105. doi: 10.1186/s40164-023-00466-3. Exp Hematol Oncol. 2023. PMID: 38104151 Free PMC article.
-
Targeting SKA3 suppresses the proliferation and chemoresistance of laryngeal squamous cell carcinoma via impairing PLK1-AKT axis-mediated glycolysis.Cell Death Dis. 2020 Oct 26;11(10):919. doi: 10.1038/s41419-020-03104-6. Cell Death Dis. 2020. PMID: 33106477 Free PMC article.
-
Glutathione levels are associated with methotrexate resistance in acute lymphoblastic leukemia cell lines.Front Oncol. 2022 Dec 1;12:1032336. doi: 10.3389/fonc.2022.1032336. eCollection 2022. Front Oncol. 2022. PMID: 36531023 Free PMC article.
-
Plasma cell dependence on histone/protein deacetylase 11 reveals a therapeutic target in multiple myeloma.JCI Insight. 2021 Dec 22;6(24):e151713. doi: 10.1172/jci.insight.151713. JCI Insight. 2021. PMID: 34793338 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
