

α1-antitrypsin mitigates NLRP3-inflammasome activation in amyloid β1-42-stimulated murine astrocytes
- PMID: 30261895
- PMCID: PMC6158809
- DOI: 10.1186/s12974-018-1319-x
α1-antitrypsin mitigates NLRP3-inflammasome activation in amyloid β1-42-stimulated murine astrocytes
Abstract
Background: Neuroinflammation has an essential impact on the pathogenesis and progression of Alzheimer's disease (AD). Mostly mediated by microglia and astrocytes, inflammatory processes lead to degeneration of neuronal cells. The NLRP3-inflammasome (NOD-like receptor family, pyrin domain containing 3) is a key component of the innate immune system and its activation results in secretion of the proinflammatory effectors interleukin-1β (IL-1β) and interleukin-18 (IL-18). Under physiological conditions, cytosolic NLRP3-inflammsome is maintained in an inactive form, not able to oligomerize. Amyloid β1-42 (Aβ1-42) triggers activation of NLRP3-inflammasome in microglia and astrocytes, inducing oligomerization and thus recruitment of proinflammatory proteases. NLRP3-inflammasome was found highly expressed in human brains diagnosed with AD. Moreover, NLRP3-deficient mice carrying mutations associated with familial AD were partially protected from deficits associated with AD. The endogenous protease inhibitor α1-antitrypsin (A1AT) is known for its anti-inflammatory and anti-apoptotic properties and thus could serve as therapeutic agent for NLRP3-inhibition. A1AT protects neurons from glutamate-induced toxicity and reduces Aβ1-42-induced inflammation in microglial cells. In this study, we investigated the effect of Aβ1-42-induced NLRP3-inflammasome upregulation in primary murine astrocytes and its regulation by A1AT.
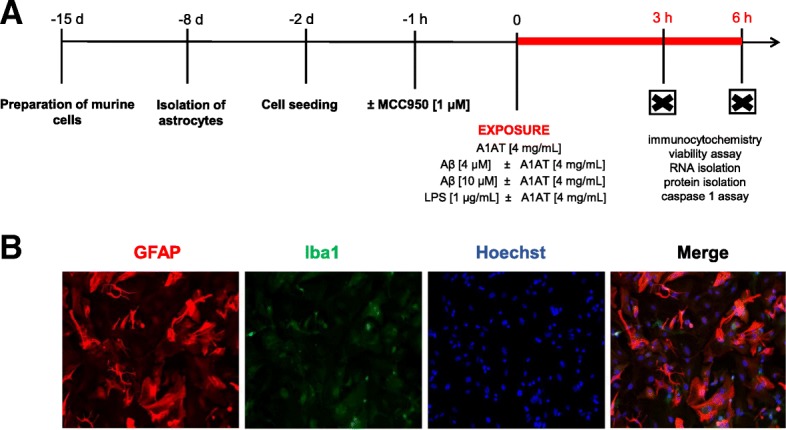
Methods: Primary cortical astrocytes from BALB/c mice were stimulated with Aβ1-42 and treated with A1AT. Regulation of NLRP3-inflammasome was examined by immunocytochemistry, PCR, western blot and ELISA. Our studies included an inhibitor of NLRP3 to elucidate direct interactions between A1AT and NLRP3-inflammasome components.
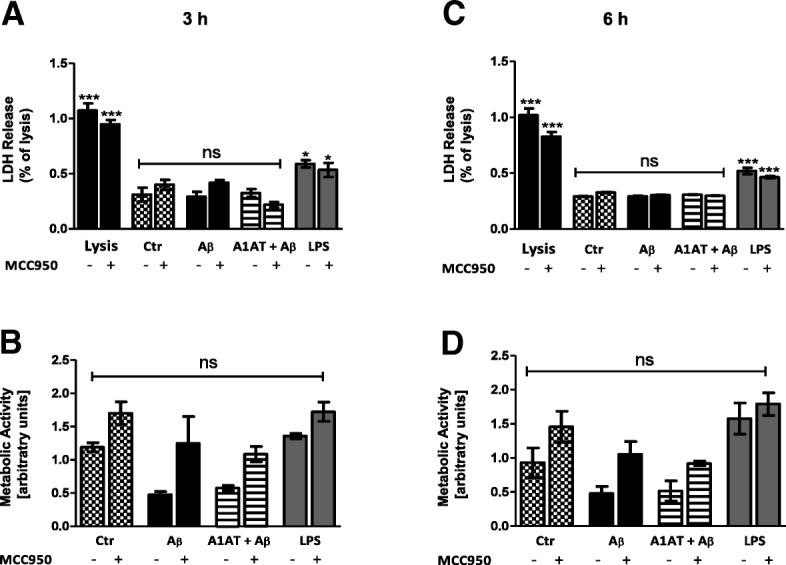
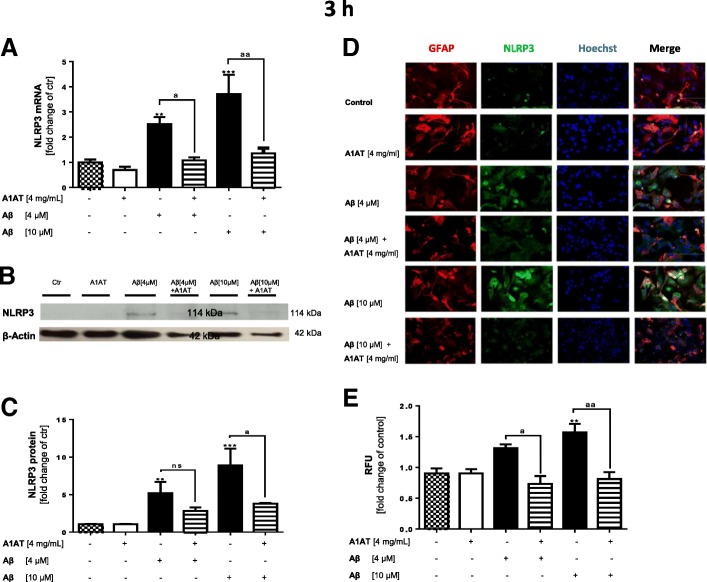
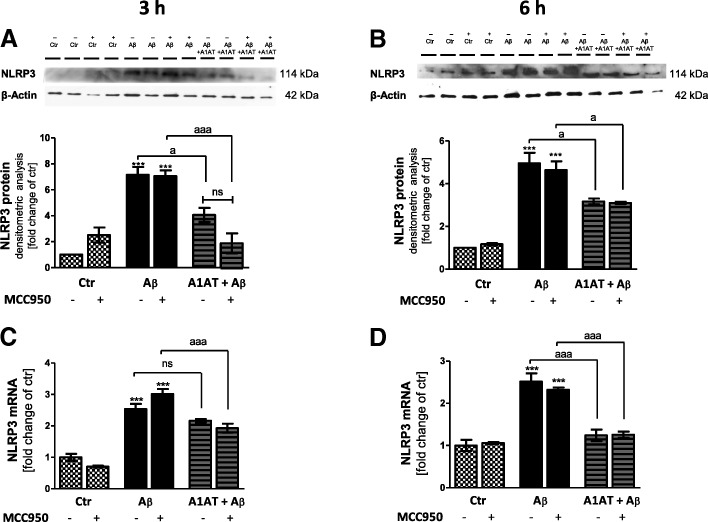
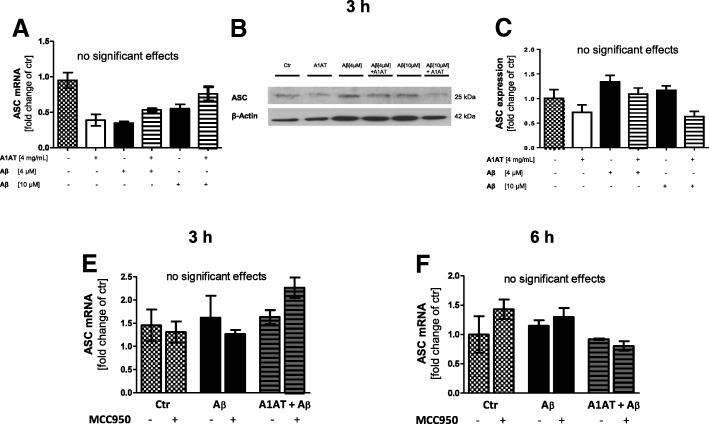
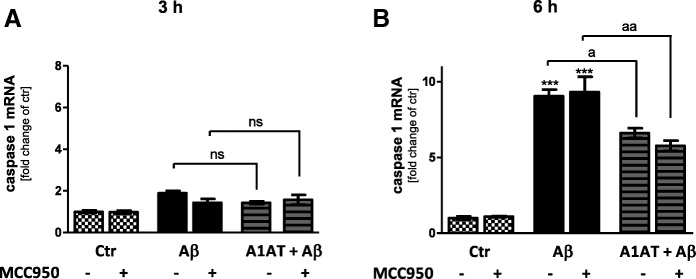
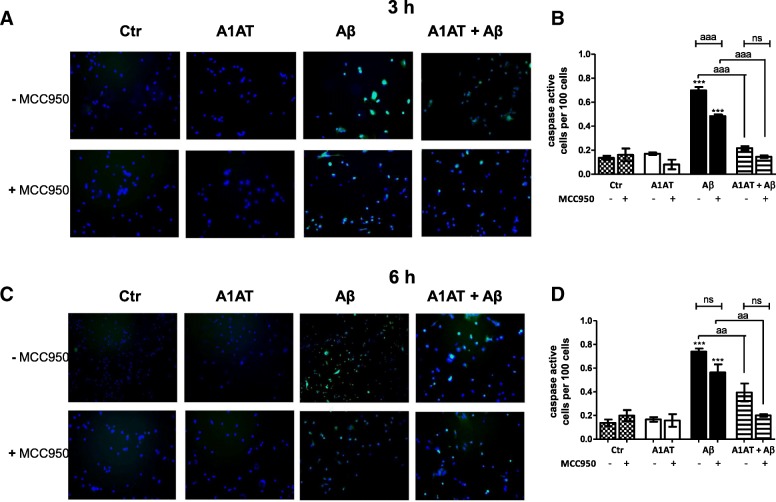
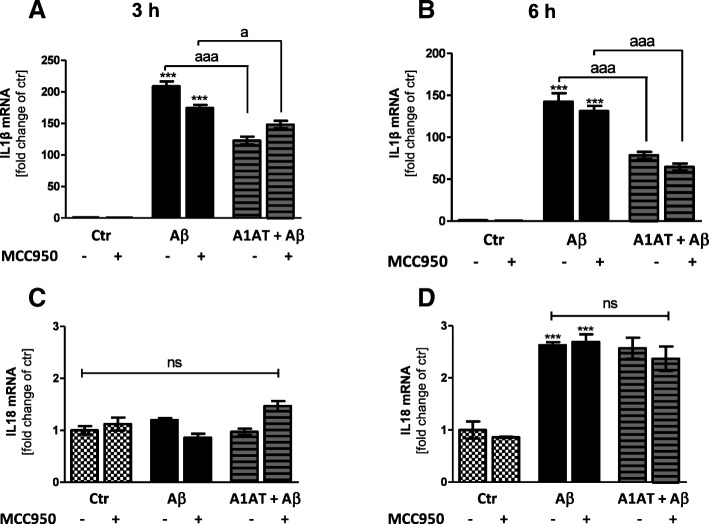
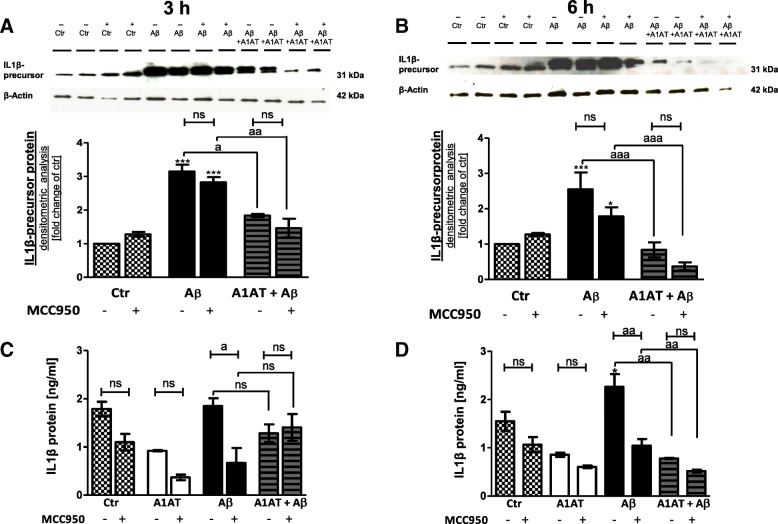
Results: Our study revealed that A1AT reduces Aβ1-42-dependent upregulation of NLRP3 at the mRNA and protein levels. Furthermore, A1AT time-dependently mitigated the expression of caspase 1 and its cleavage product IL-1β in Aβ1-42-stimulated astrocytes.
Conclusion: We conclude that Aβ1-42-stimulation results in an upregulation of NLRP3, caspase 1, and its cleavage products in astrocytes. A1AT time-dependently hampers neuroinflammation by downregulation of Aβ1-42-mediated NLRP3-inflammasome expression and thus may serve as a pharmaceutical opportunity for the treatment of Alzheimer's disease.
Keywords: Alpha 1-antitrypsin; Alzheimer’s disease; Amyloid β; Astrocytes; Inflammasome; NALP3; NLRP3; Neuroinflammation.
Conflict of interest statement
Ethics approval
No human tissue was involved in this study. Postnatal (P0 to P2) cortical astrocyte culture preparation from BALB/c mice (Charles River) was performed as previously described by Habib et al. 2014 [51]. Preparation was conducted in accordance with animal welfare policy of University Hospital Aachen and the government of the State of North Rhine-Westphalia, Germany (no. 84.02.04.2015.A292).
Consent for publication
Not applicable
Competing interests
All authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
