

Nanoscale electrochemical kinetics & dynamics: the challenges and opportunities of single-entity measurements
- PMID: 30264833
- PMCID: PMC6261779
- DOI: 10.1039/c8fd00134k
Nanoscale electrochemical kinetics & dynamics: the challenges and opportunities of single-entity measurements
Abstract
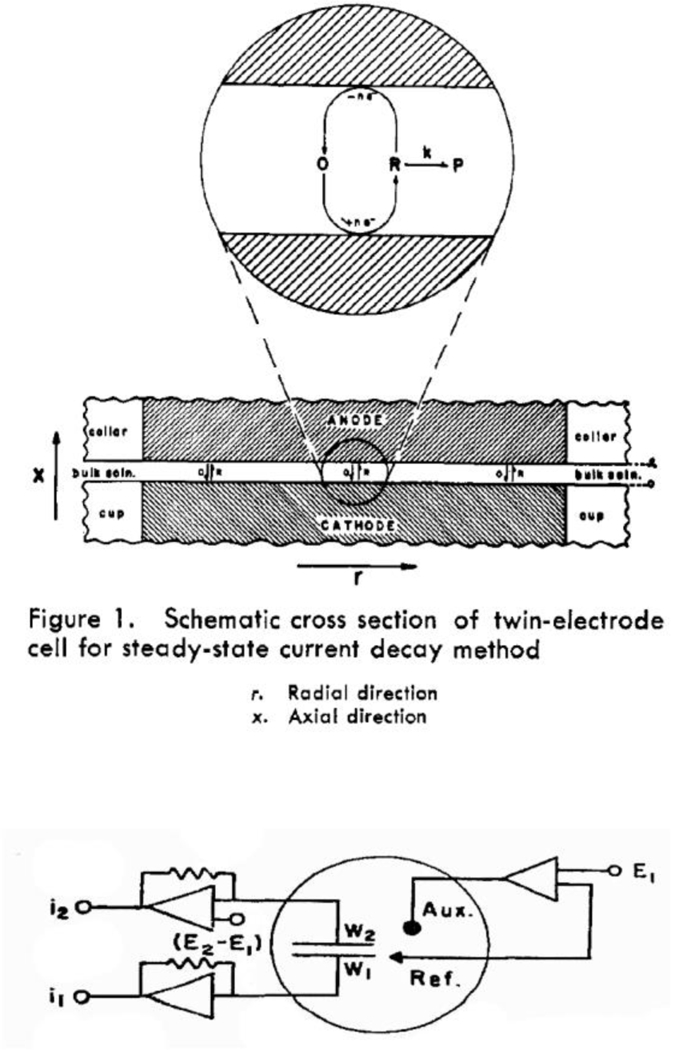
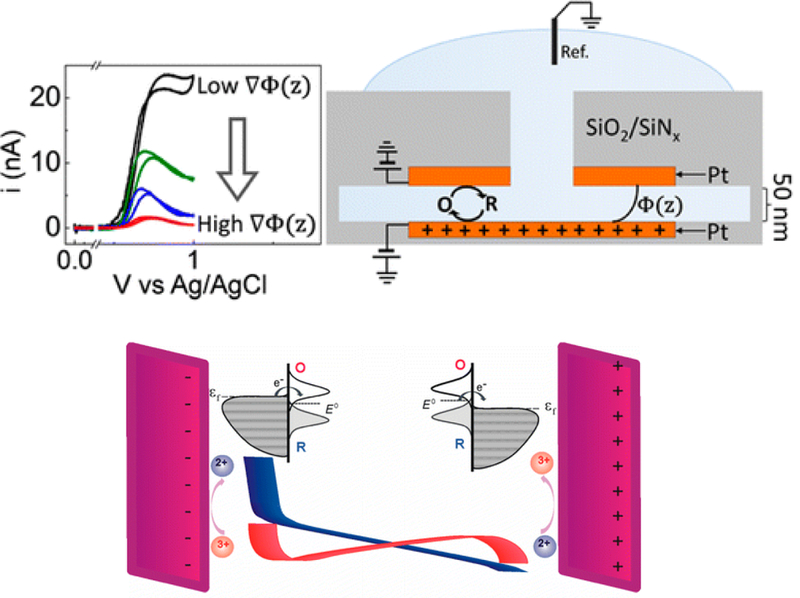
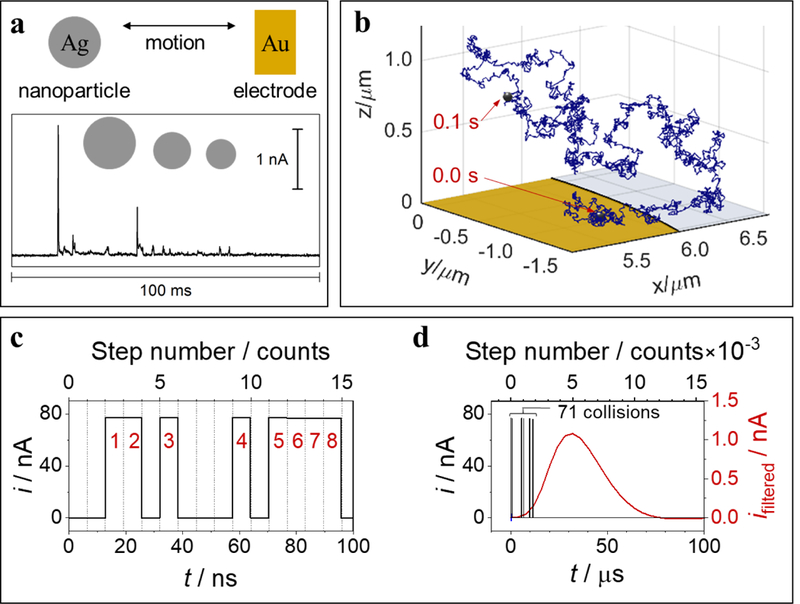
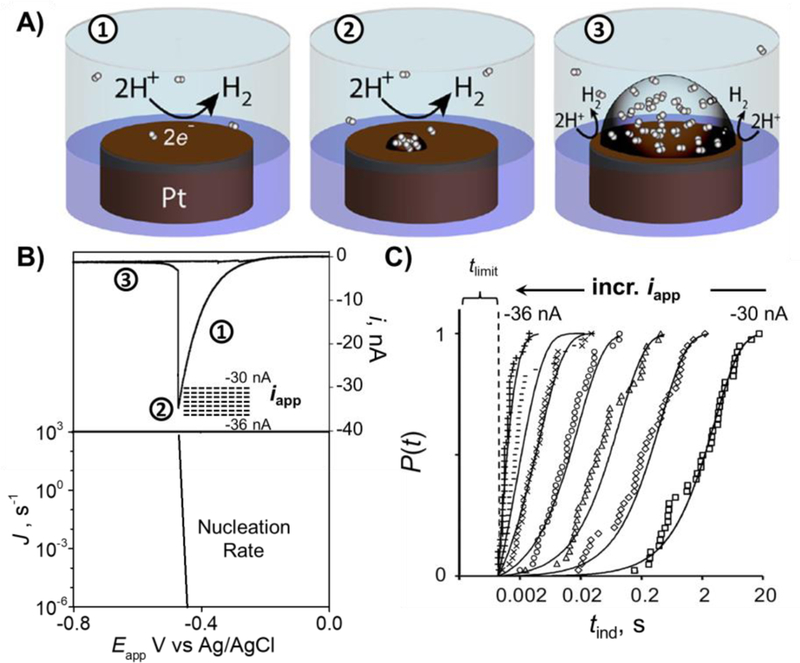
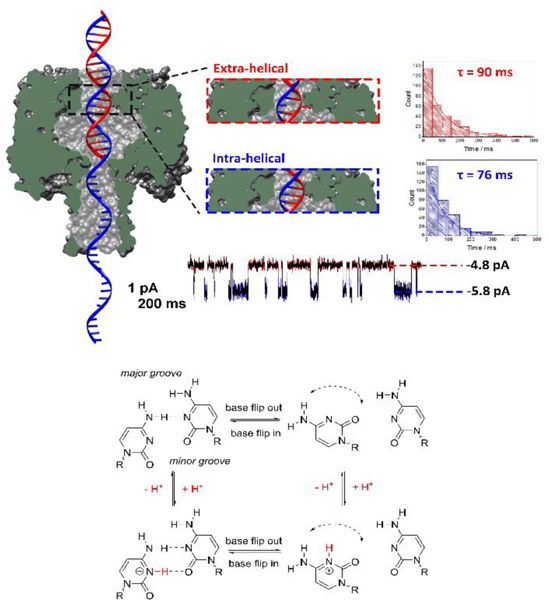
The development of nanoscale electrochemistry since the mid-1980s has been predominately coupled with steady-state voltammetric (i-E) methods. This research has been driven by the desire to understand the mechanisms of very fast electrochemical reactions, by electroanalytical measurements in small volumes and unusual media, including in vivo measurements, and by research on correlating electrocatalytic activity, e.g., O2 reduction reaction, with nanoparticle size and structure. Exploration of the behavior of nanoelectrochemical structures (nanoelectrodes, nanoparticles, nanogap cells, etc.) of a characteristic dimension λ using steady-state i-E methods generally relies on the well-known relationship, λ2 ∼ Dt, which relates diffusional lengths to time, t, through the coefficient, D. Decreasing λ, by performing measurements at a nanometric length scales, results in a decrease in the effective timescale of the measurement, and provides a direct means to probe the kinetics of steps associated with very rapid electrochemical reactions. For instance, steady-state voltammetry using a nanogap twin-electrode cell of characteristic width, λ ∼ 10 nm, allows investigations of events occurring at timescales on the order of ∼100 ns. Among many other advantages, decreasing λ also increases spatial resolution in electrochemical imaging, e.g., in scanning electrochemical microscopy, and allows probing of the electric double layer. This Introductory Lecture traces the evolution and driving forces behind the "λ2 ∼ Dt" steady-state approach to nanoscale electrochemistry, beginning in the late 1950s with the introduction of the rotating ring-disk electrode and twin-electrode thin-layer cells, and evolving to current-day investigations using nanoelectrodes, scanning nanocells for imaging, nanopores, and nanoparticles. The recent focus on so-called "single-entity" electrochemistry, in which individual and very short redox events are probed, is a significant departure from the steady-state approach, but provides new opportunities to probe reaction dynamics. The stochastic nature of very fast single-entity events challenges current electrochemical methods and modern electronics, as illustrated using recent experiments from the authors' laboratory.
Figures


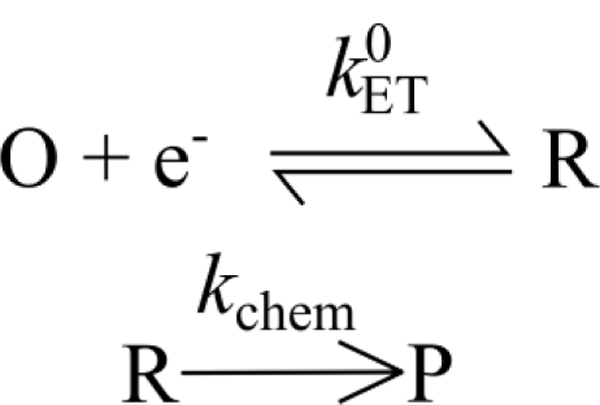
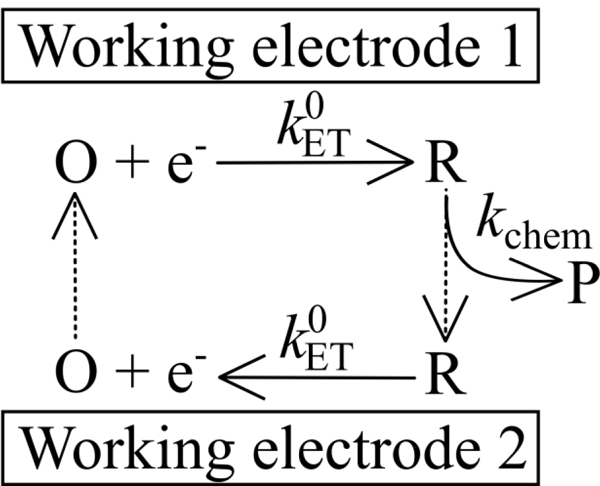
References
-
- Murray RW, Chem. Rev, 2008, 108, 2688–2720. - PubMed
-
- Oja SM, Fan YS, Armstrong CM, Defnet P and Zhang B, Anal. Chem, 2016, 88, 414–430. - PubMed
-
- Katemann BB and Schuhmann W, Electroanalysis, 2002, 14, 22–28.
-
- Yu Y, Sun T and Mirkin MV, Anal. Chem, 2016, 88, 11758–11766. - PubMed
-
- Wang W, Zhang J, Wang FF, Mao BW, Zhan DP and Tian ZQ, J. Am. Chem. Soc, 2016, 138, 9057–9060. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous