

Nonclassical GH Insensitivity: Characterization of Mild Abnormalities of GH Action
- PMID: 30265312
- PMCID: PMC6607971
- DOI: 10.1210/er.2018-00146
Nonclassical GH Insensitivity: Characterization of Mild Abnormalities of GH Action
Abstract
GH insensitivity (GHI) presents in childhood with growth failure and in its severe form is associated with extreme short stature and dysmorphic and metabolic abnormalities. In recent years, the clinical, biochemical, and genetic characteristics of GHI and other overlapping short stature syndromes have rapidly expanded. This can be attributed to advancing genetic techniques and a greater awareness of this group of disorders. We review this important spectrum of defects, which present with phenotypes at the milder end of the GHI continuum. We discuss their clinical, biochemical, and genetic characteristics. The objective of this review is to clarify the definition, identification, and investigation of this clinically relevant group of growth defects. We also review the therapeutic challenges of mild GHI.
Copyright © 2019 Endocrine Society.
Figures
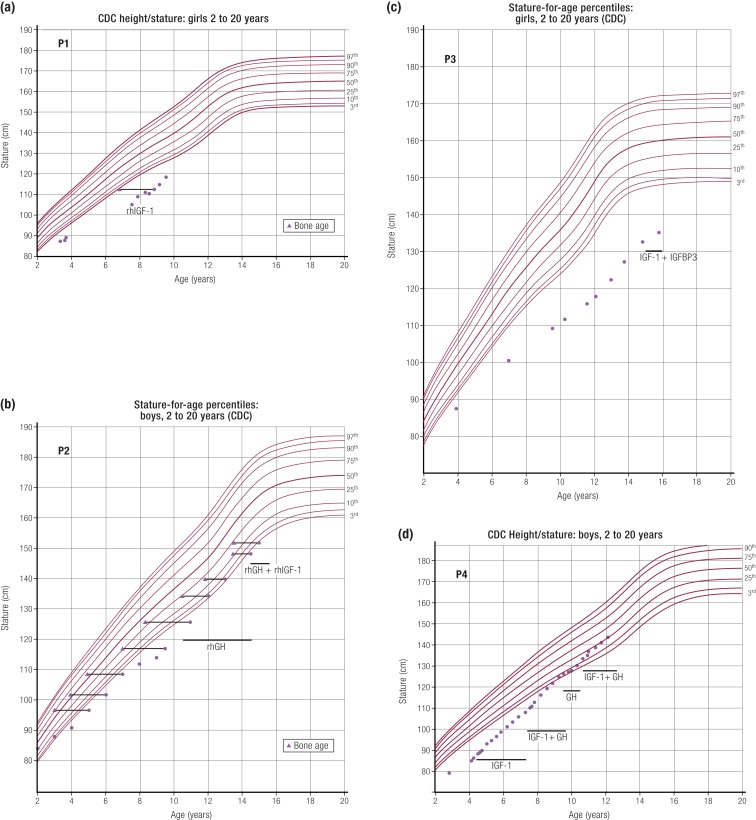
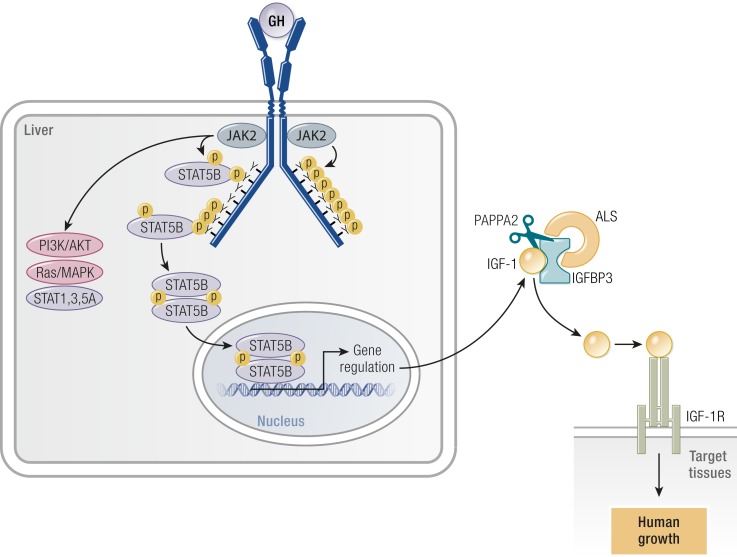
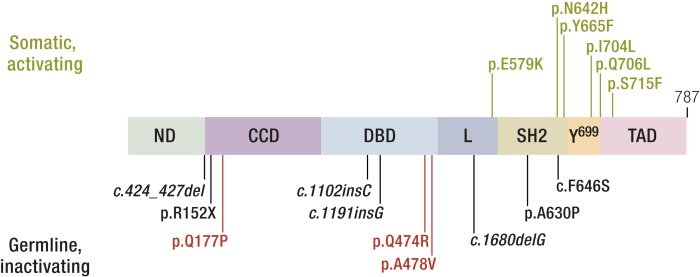
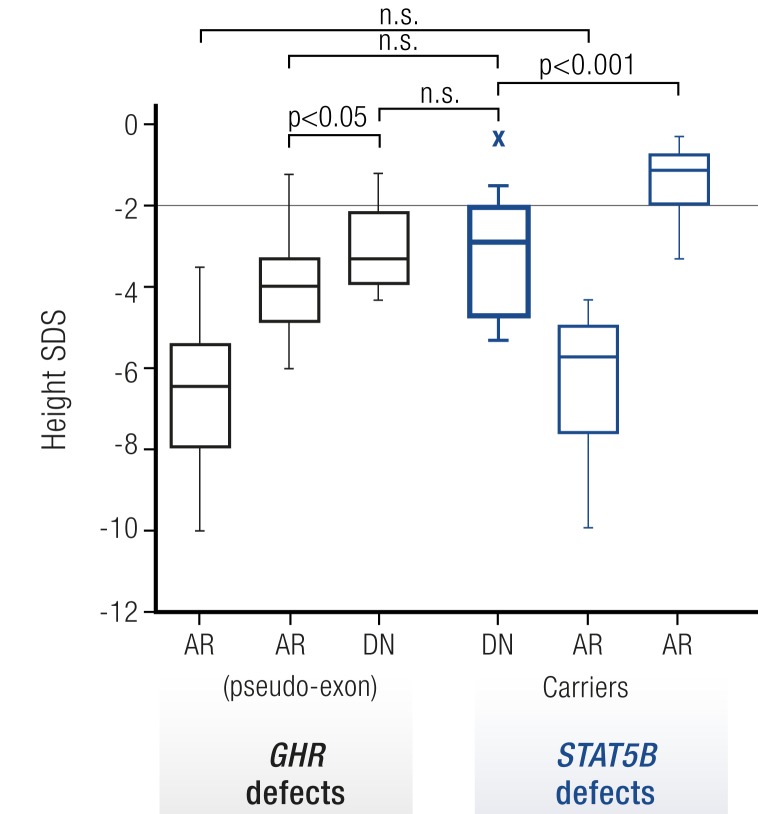
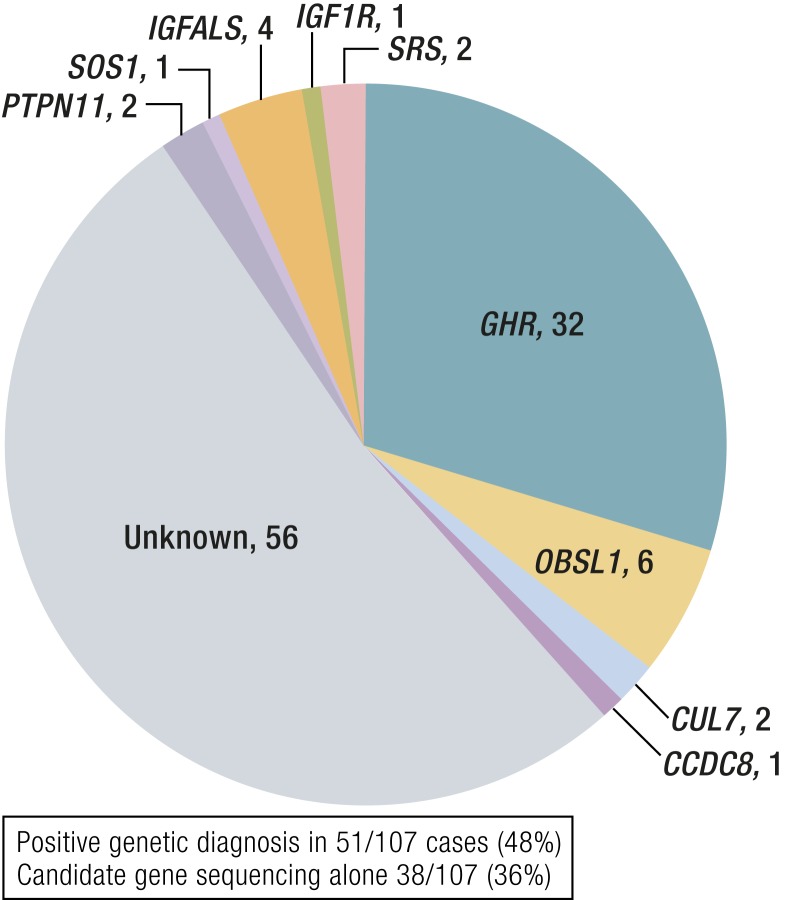
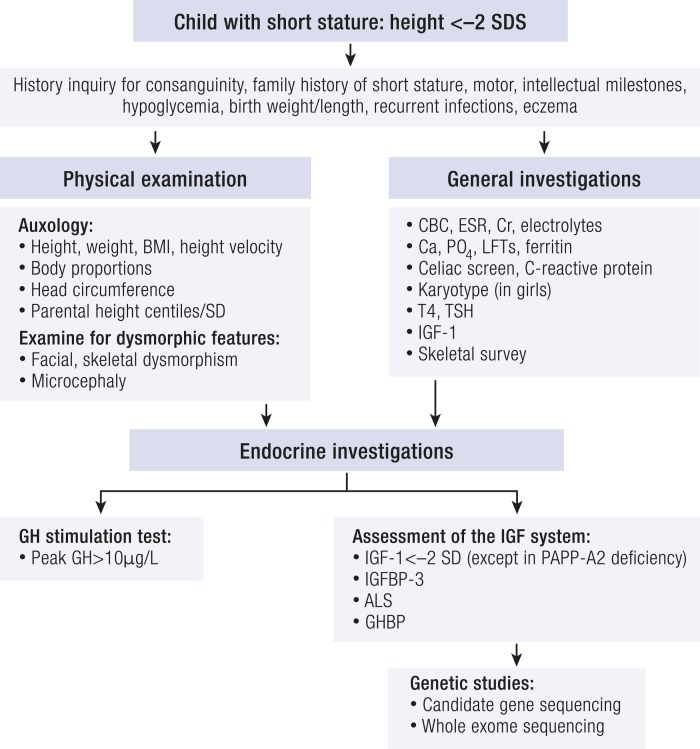
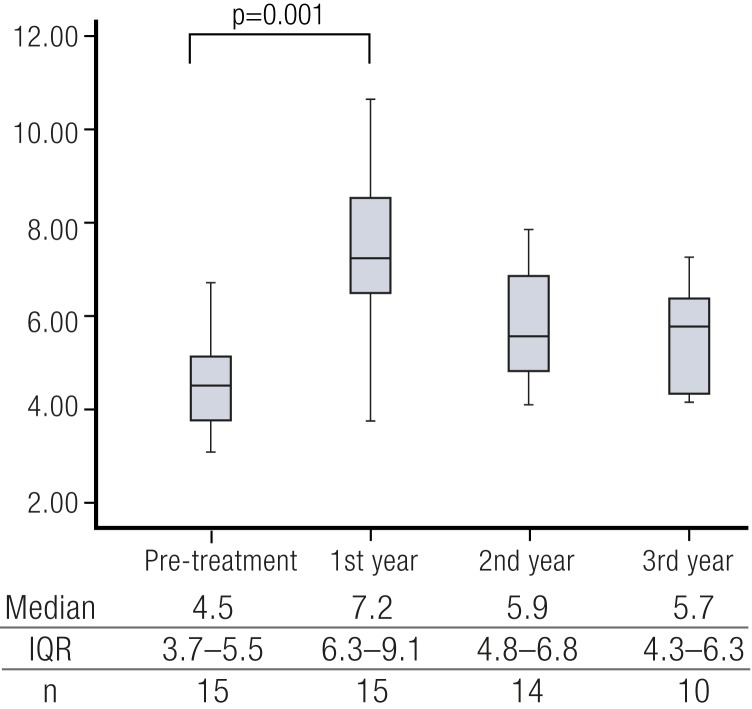
References
-
- David A, Hwa V, Metherell LA, Netchine I, Camacho-Hübner C, Clark AJ, Rosenfeld RG, Savage MO. Evidence for a continuum of genetic, phenotypic, and biochemical abnormalities in children with growth hormone insensitivity. Endocr Rev. 2011;32(4):472–497. - PubMed
-
- Laron Z. Laron syndrome (primary growth hormone resistance or insensitivity): the personal experience 1958–2003. J Clin Endocrinol Metab. 2004;89(3):1031–1044. - PubMed
-
- Laron Z, Pertzelan A, Mannheimer S. Genetic pituitary dwarfism with high serum concentation of growth hormone—a new inborn error of metabolism? Isr J Med Sci. 1966;2(2):152–155. - PubMed
-
- Eshet R, Laron Z, Pertzelan A, Arnon R, Dintzman M. Defect of human growth hormone receptors in the liver of two patients with Laron-type dwarfism. Isr J Med Sci. 1984;20(1):8–11. - PubMed
-
- Savage MO, Burren CP, Rosenfeld RG. The continuum of growth hormone–IGF-I axis defects causing short stature: diagnostic and therapeutic challenges. Clin Endocrinol (Oxf). 2010;72(6):721–728. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
