

Mechanism-based rescue of Munc18-1 dysfunction in varied encephalopathies by chemical chaperones
- PMID: 30266908
- PMCID: PMC6162227
- DOI: 10.1038/s41467-018-06507-4
Mechanism-based rescue of Munc18-1 dysfunction in varied encephalopathies by chemical chaperones
Abstract
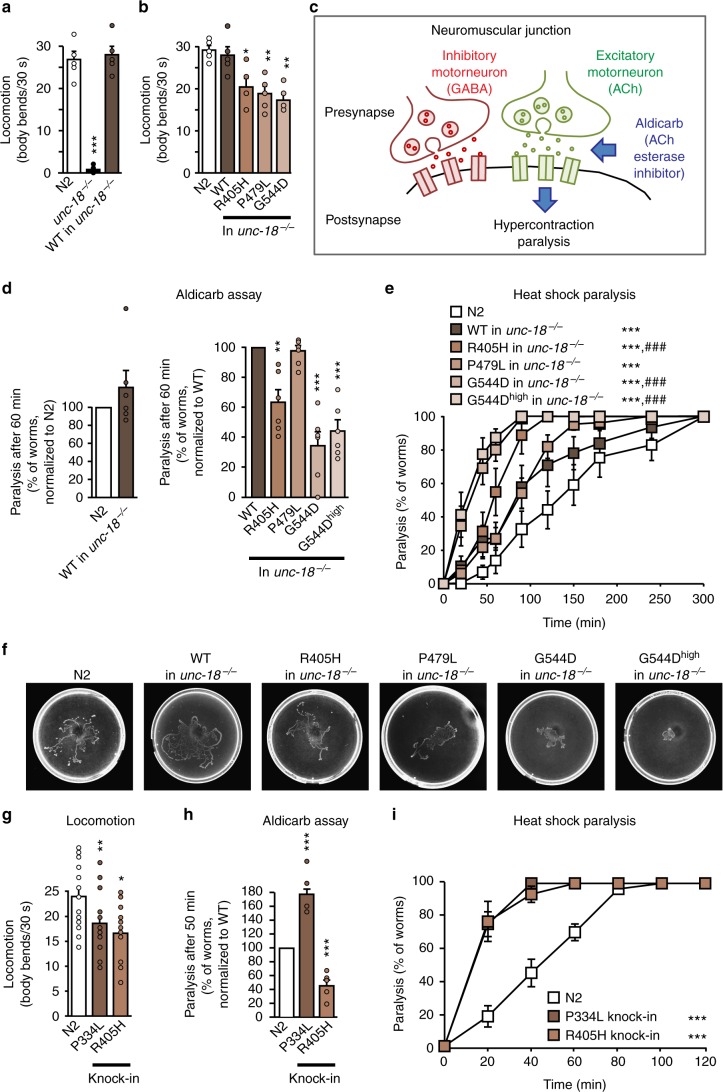
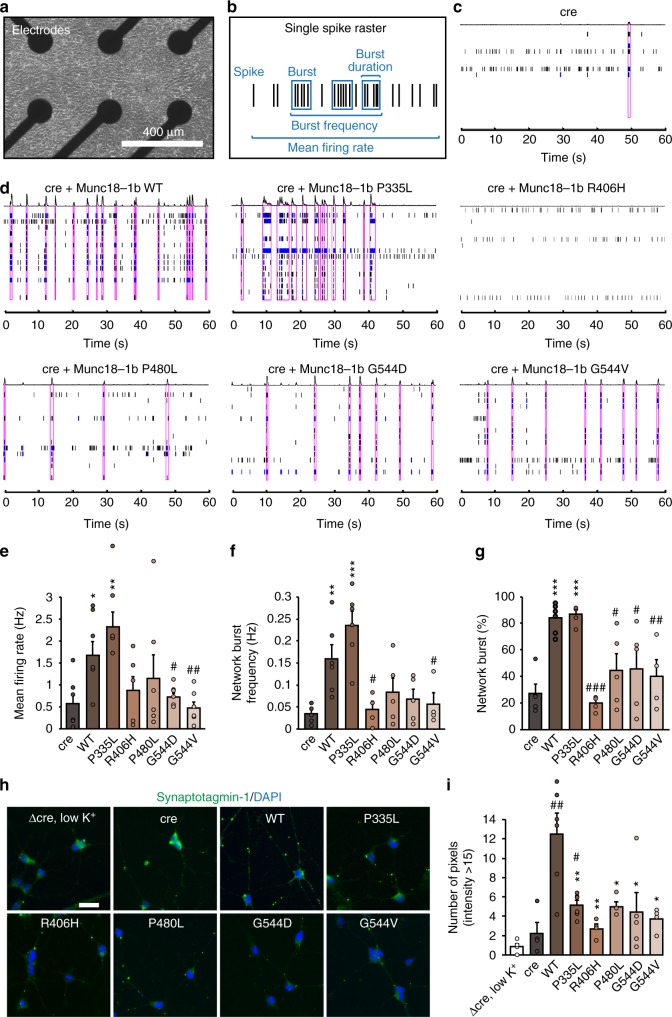
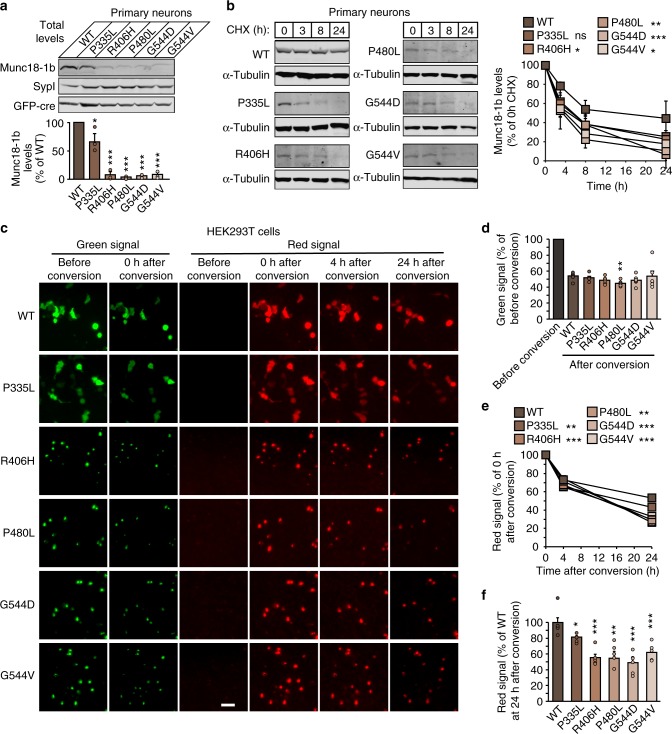
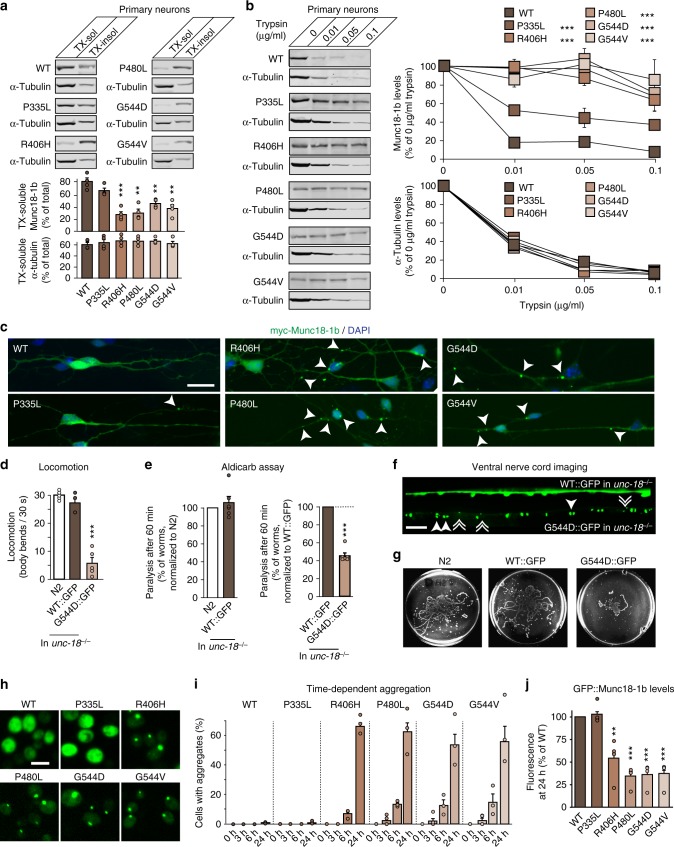
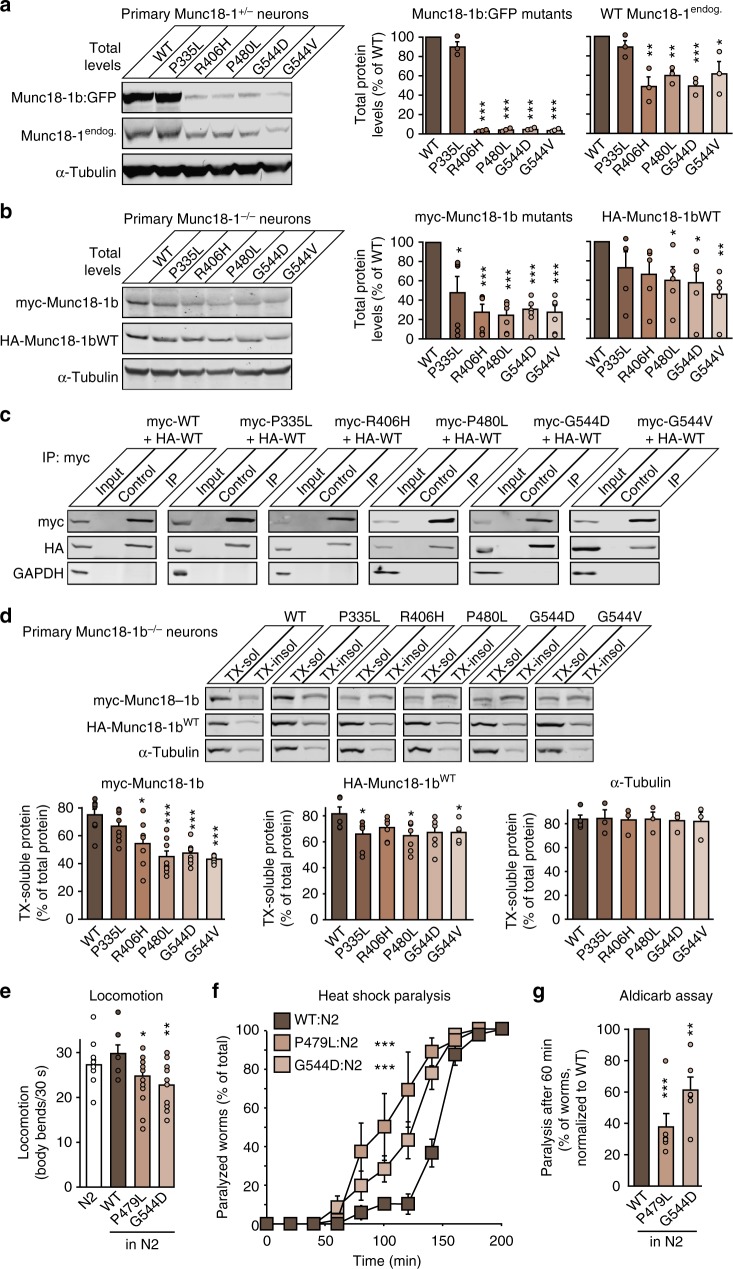
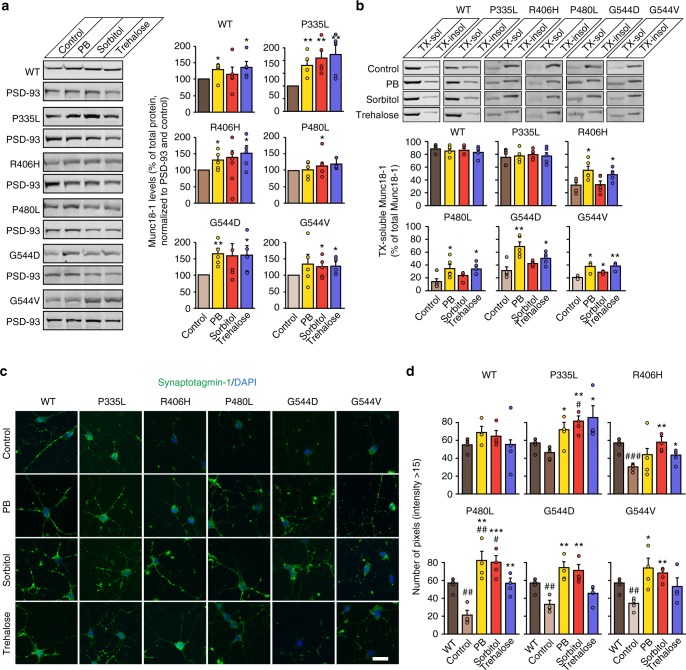
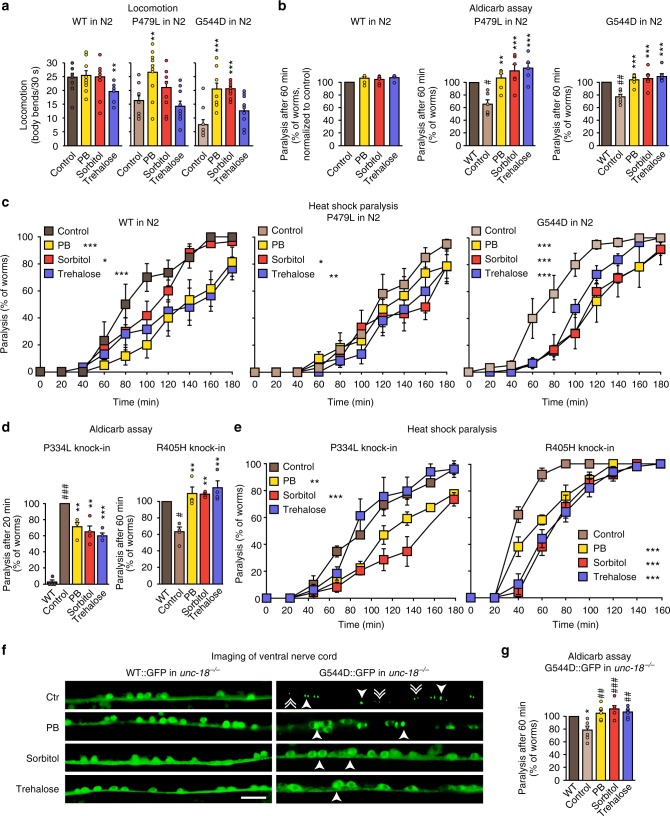
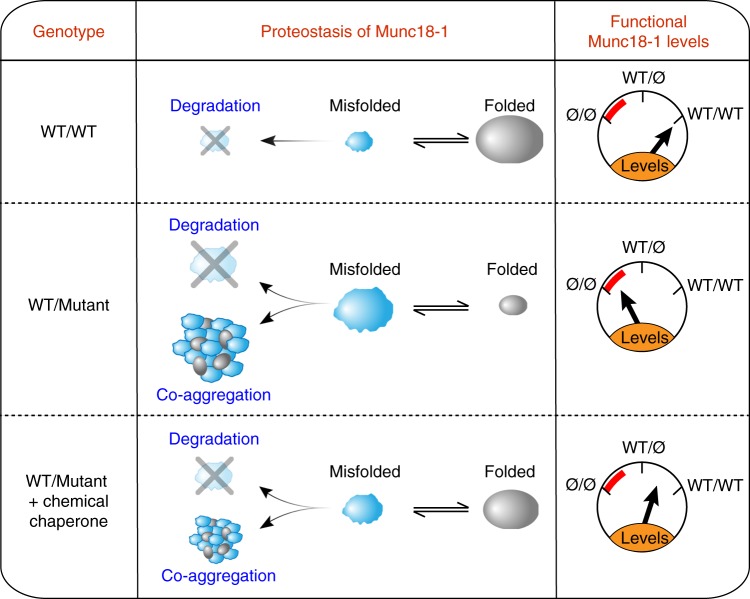
Heterozygous de novo mutations in the neuronal protein Munc18-1 are linked to epilepsies, intellectual disability, movement disorders, and neurodegeneration. These devastating diseases have a poor prognosis and no known cure, due to lack of understanding of the underlying disease mechanism. To determine how mutations in Munc18-1 cause disease, we use newly generated S. cerevisiae strains, C. elegans models, and conditional Munc18-1 knockout mouse neurons expressing wild-type or mutant Munc18-1, as well as in vitro studies. We find that at least five disease-linked missense mutations of Munc18-1 result in destabilization and aggregation of the mutant protein. Aggregates of mutant Munc18-1 incorporate wild-type Munc18-1, depleting functional Munc18-1 levels beyond hemizygous levels. We demonstrate that the three chemical chaperones 4-phenylbutyrate, sorbitol, and trehalose reverse the deficits caused by mutations in Munc18-1 in vitro and in vivo in multiple models, offering a novel strategy for the treatment of varied encephalopathies.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- R01-GM095674/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)/International
- R01 GM095674/GM/NIGMS NIH HHS/United States
- T32GM007739/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)/International
- R01 NS102181/NS/NINDS NIH HHS/United States
- T32 GM007739/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
