

Insights From Deep Sequencing of the HBV Genome-Unique, Tiny, and Misunderstood
- PMID: 30268787
- PMCID: PMC6347571
- DOI: 10.1053/j.gastro.2018.07.058
Insights From Deep Sequencing of the HBV Genome-Unique, Tiny, and Misunderstood
Abstract
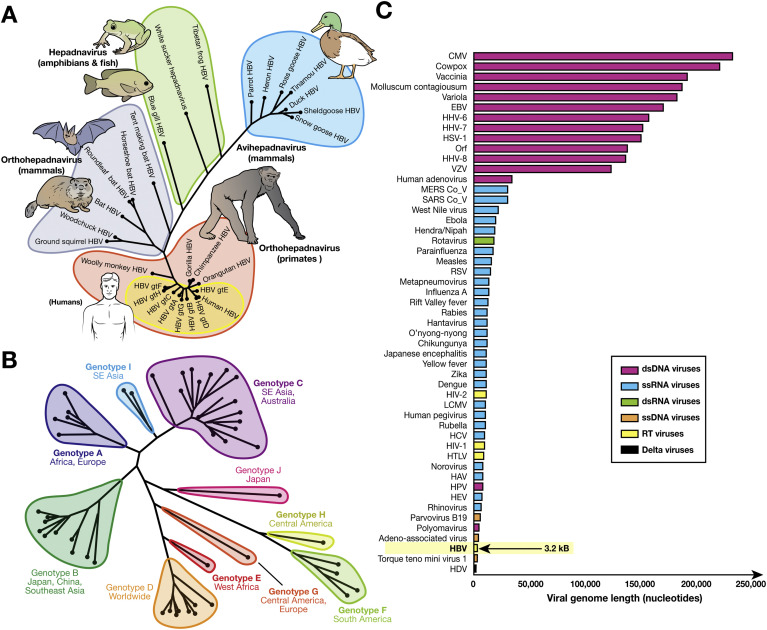
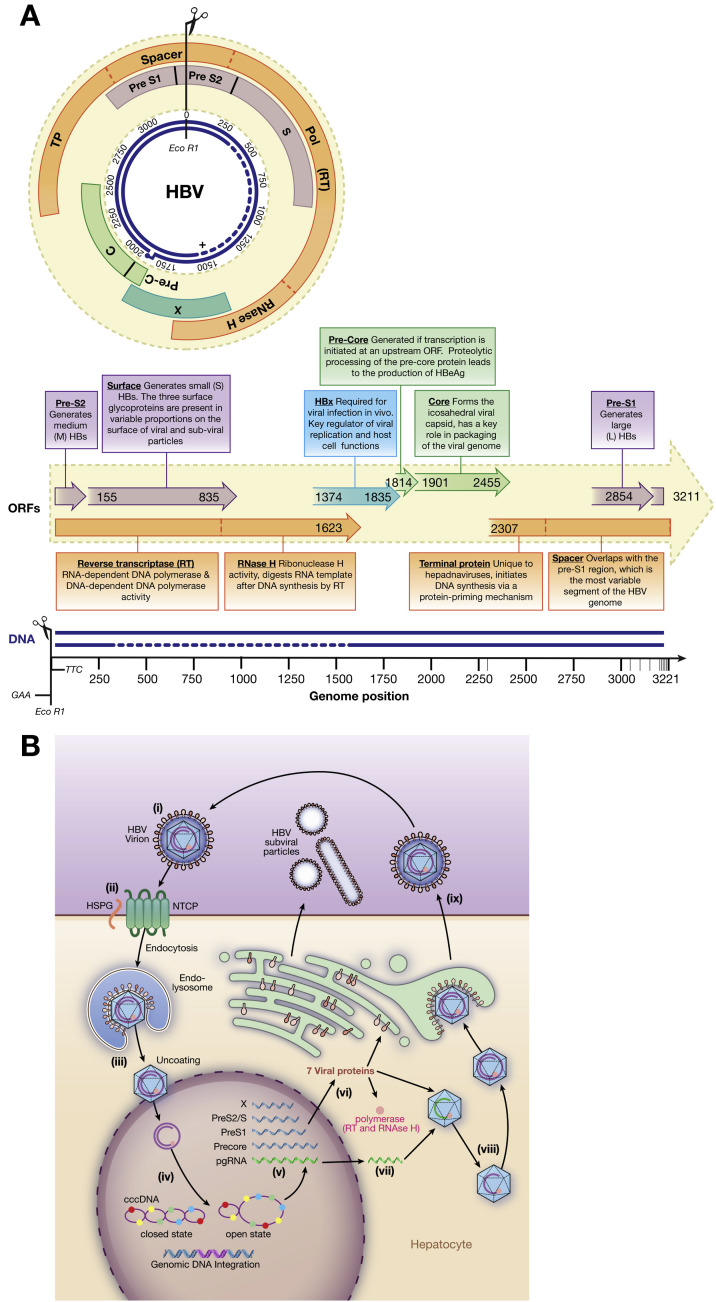
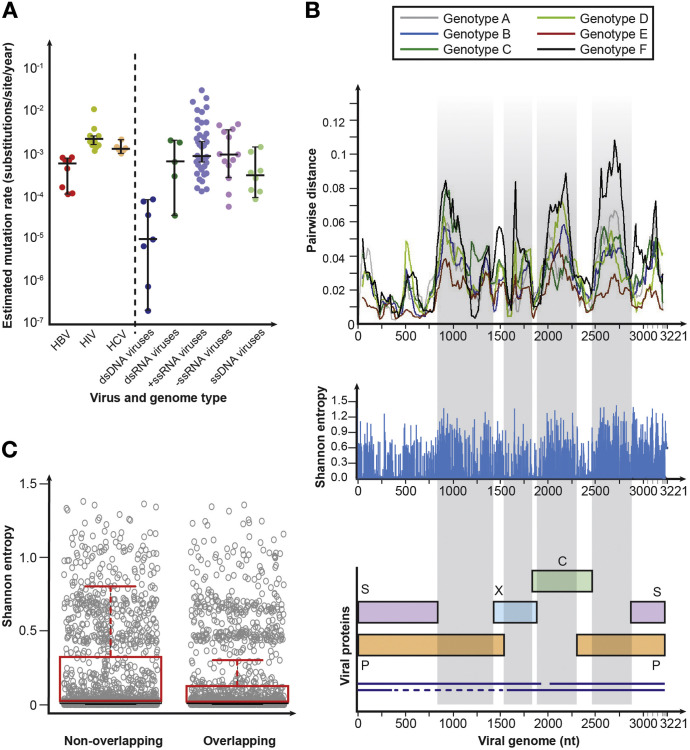
Hepatitis B virus (HBV) is a unique, tiny, partially double-stranded, reverse-transcribing DNA virus with proteins encoded by multiple overlapping reading frames. The substitution rate is surprisingly high for a DNA virus, but lower than that of other reverse transcribing organisms. More than 260 million people worldwide have chronic HBV infection, which causes 0.8 million deaths a year. Because of the high burden of disease, international health agencies have set the goal of eliminating HBV infection by 2030. Nonetheless, the intriguing HBV genome has not been well characterized. We summarize data on the HBV genome structure and replication cycle, explain and quantify diversity within and among infected individuals, and discuss advances that can be offered by application of next-generation sequencing technology. In-depth HBV genome analyses could increase our understanding of disease pathogenesis and allow us to better predict patient outcomes, optimize treatment, and develop new therapeutics.
Keywords: Diversity; Evolution; Genotype; Hepatitis B Virus.
Copyright © 2019 AGA Institute. Published by Elsevier Inc. All rights reserved.
Figures








References
-
- London W.T., Sutnick A.I., Blumberg B.S. Australia antigen and acute viral hepatitis. Ann Intern Med. 1969;70:55–59. - PubMed
-
- Blumberg B.S., Alter H.J., Visnich S. A “new” antigen in leukemia sera. JAMA. 1965;191:541–546. - PubMed
-
- World Health Organization Preventing perinatal hepatitis B virus transmission : a guide for introducing and strengthening hepatitis B birth dose vaccination. http://apps.who.int/iris/bitstream/10665/208278/1/ Available at: Published 2015.
-
- Schweitzer A., Horn J., Mikolajczyk R.T. Estimations of worldwide prevalence of chronic hepatitis B virus infection: a systematic review of data published between 1965 and 2013. Lancet. 2015;386:1546–1555. - PubMed
-
- Matthews P.C., Geretti A.M., Goulder P.J.R. Epidemiology and impact of HIV coinfection with hepatitis B and hepatitis C viruses in Sub-Saharan Africa. J Clin Virol. 2014;61:20–33. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
