

Inhibition of Casein Kinase 2 Protects Oligodendrocytes From Excitotoxicity by Attenuating JNK/p53 Signaling Cascade
- PMID: 30271323
- PMCID: PMC6146035
- DOI: 10.3389/fnmol.2018.00333
Inhibition of Casein Kinase 2 Protects Oligodendrocytes From Excitotoxicity by Attenuating JNK/p53 Signaling Cascade
Abstract
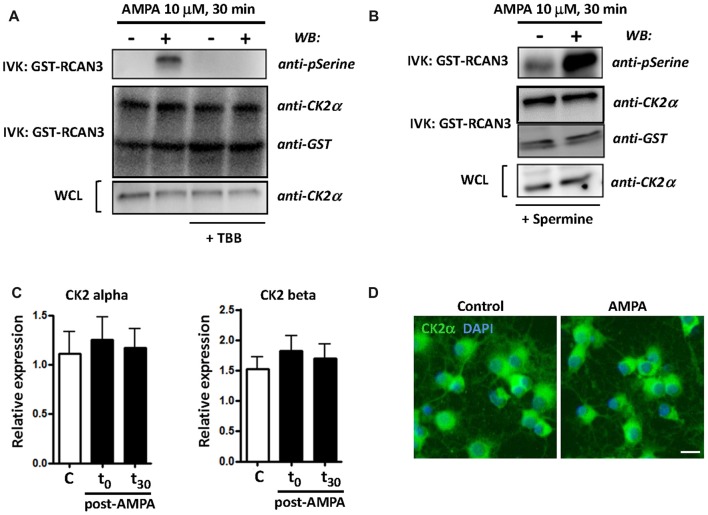
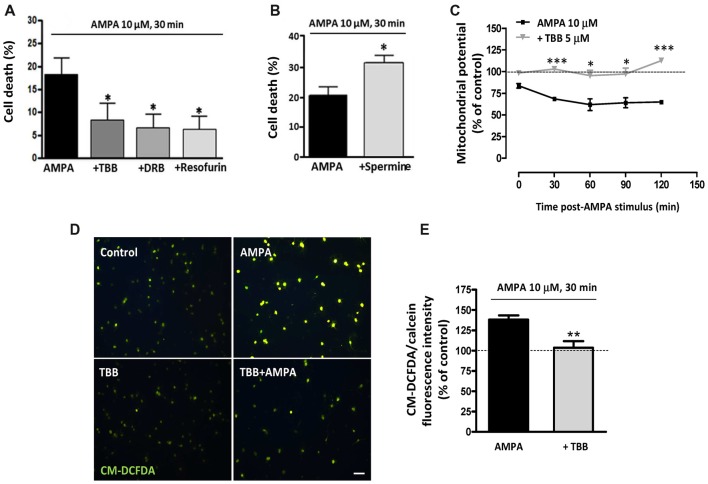
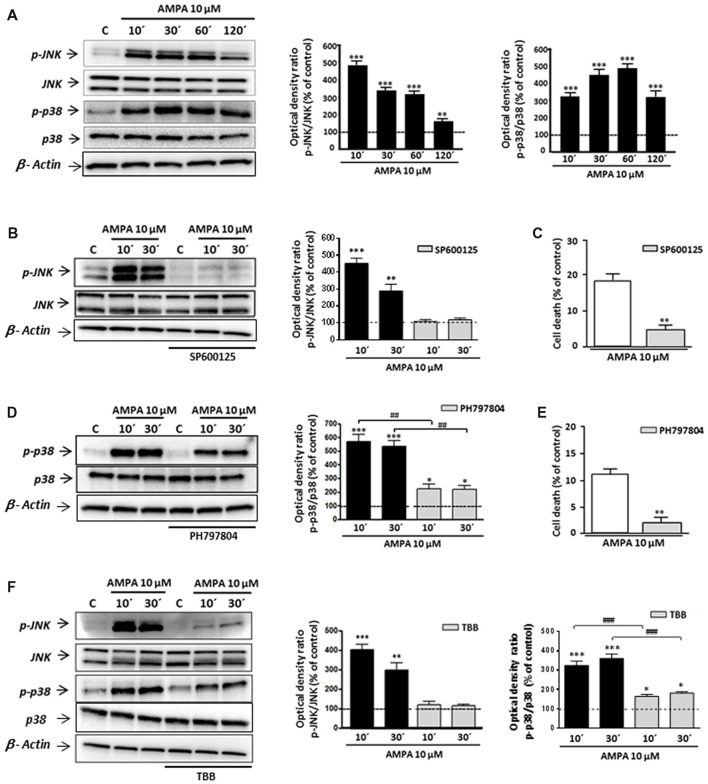
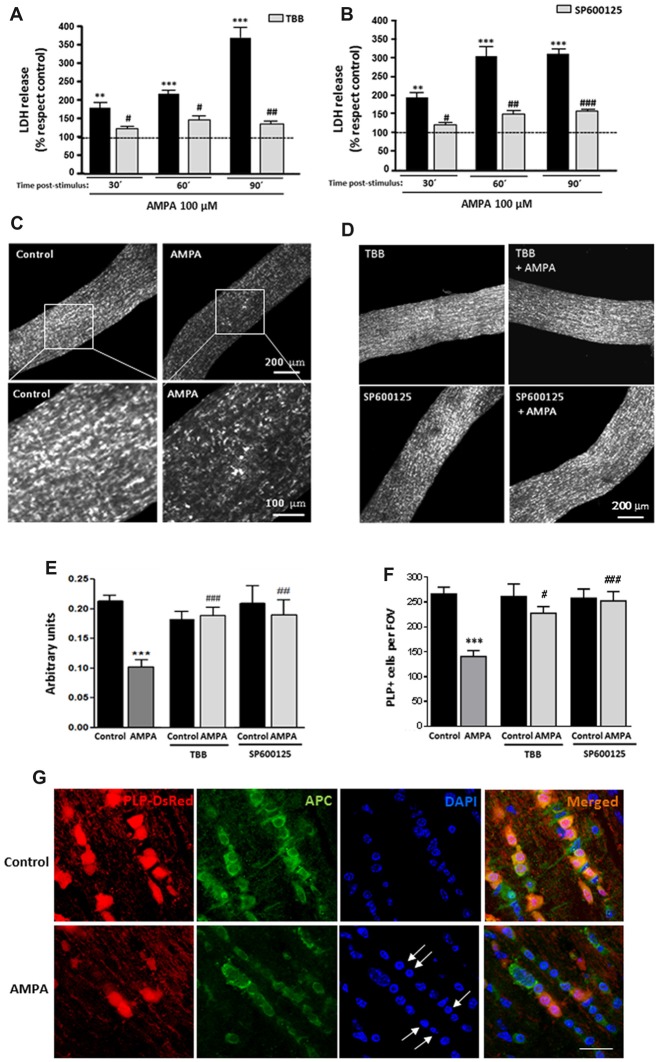
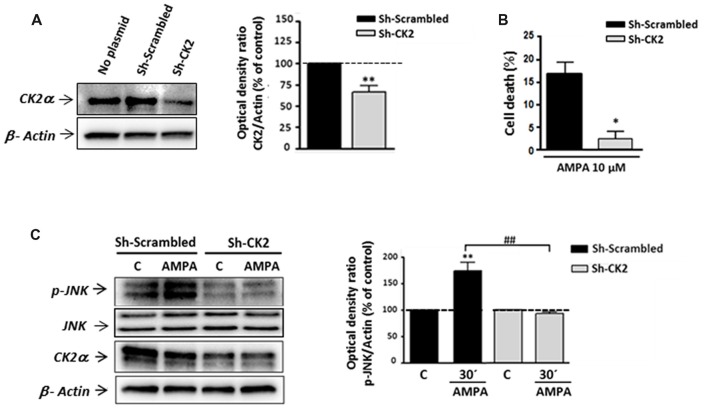
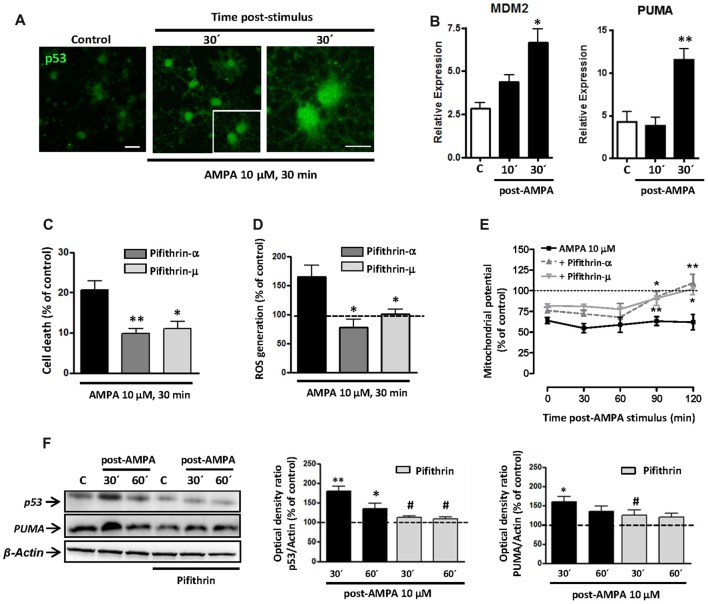
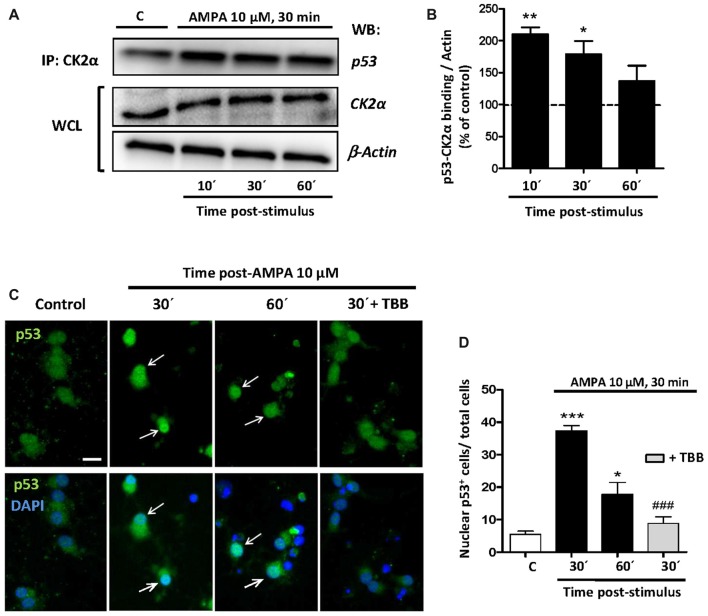
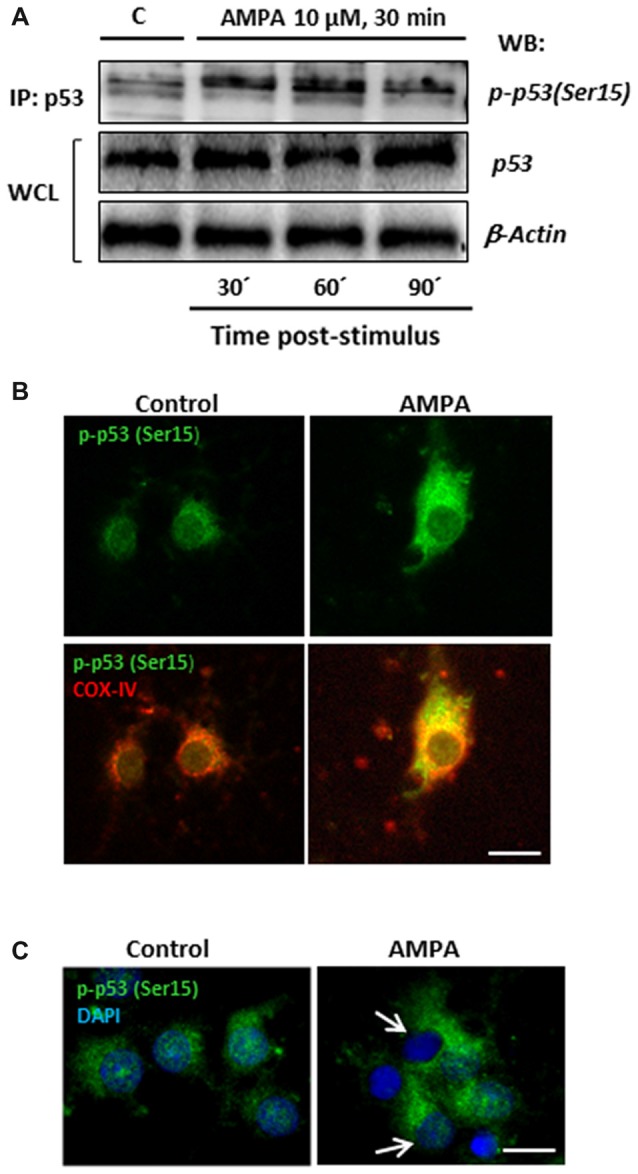
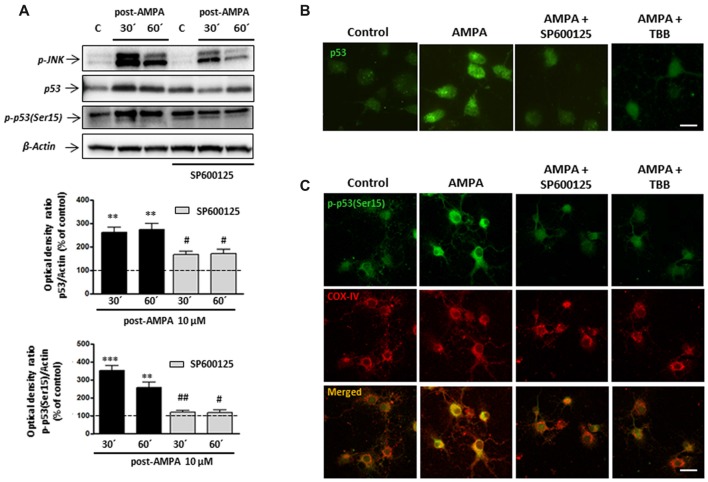
Oligodendrocytes are highly vulnerable to glutamate excitotoxicity, a central mechanism involved in tissue damage in Multiple Sclerosis (MS). Sustained activation of AMPA receptors in rat oligodendrocytes induces cytosolic calcium overload, mitochondrial depolarization, increase of reactive oxygen species, and activation of intracelular pathways resulting in apoptotic cell death. Although many signals driven by excitotoxicity have been identified, some of the key players are still under investigation. Casein kinase 2 (CK2) is a serine/threonine kinase, constitutively expressed in all eukaryotic tissues, involved in cell proliferation, malignant transformation and apoptosis. In this study, we identify CK2 as a critical regulator of oligodendrocytic death pathways and elucidate its role as a signal inductor following excitotoxic insults. We provide evidence that CK2 activity is up-regulated in AMPA-treated oligodendrocytes and CK2 inhibition significantly diminished AMPA receptor-induced oligodendroglial death. In addition, we analyzed mitogen-activated protein kinase (MAPK) signaling after excitotoxic insult. We observed that AMPA receptor activation induced a rapid increase in c-Jun N-terminal kinase (JNK) and p38 phosphorylation that was reduced after CK2 inhibition. Moreover, blocking their phosphorylation, we enhanced oligodendrocyte survival after excitotoxic insult. Finally, we observed that the tumor suppressor p53 is activated during AMPA receptor-induced cell death and, interestingly, down-regulated by JNK or CK2 inhibition. Together, these data indicate that the increase in CK2 activity induced by excitotoxic insults regulates MAPKs, triggers p53 activation and mediates subsequent oligodendroglial loss. Therefore, targeting CK2 may be a useful strategy to prevent oligodendrocyte death in MS and other diseases involving central nervous system (CNS) white matter.
Keywords: AMPA receptor; CK2; apoptosis; c-Jun N-terminal kinase (JNK); excitotoxicity; mitogen-activated protein kinase (MAPK); oligodendrocyte; p53.
Figures
Similar articles
-
GABA Receptor Agonists Protect From Excitotoxic Damage Induced by AMPA in Oligodendrocytes.Front Pharmacol. 2022 Jul 26;13:897056. doi: 10.3389/fphar.2022.897056. eCollection 2022. Front Pharmacol. 2022. PMID: 35959434 Free PMC article.
-
Dual-specific phosphatase-6 (Dusp6) and ERK mediate AMPA receptor-induced oligodendrocyte death.J Biol Chem. 2011 Apr 1;286(13):11825-36. doi: 10.1074/jbc.M110.153049. Epub 2011 Feb 7. J Biol Chem. 2011. PMID: 21300799 Free PMC article.
-
Pyrrolidine dithiocarbamate-induced neuronal cell death is mediated by Akt, casein kinase 2, c-Jun N-terminal kinase, and IkappaB kinase in embryonic hippocampal progenitor cells.J Neurosci Res. 2003 Mar 1;71(5):689-700. doi: 10.1002/jnr.10520. J Neurosci Res. 2003. PMID: 12584727
-
Glutamate, T cells and multiple sclerosis.J Neural Transm (Vienna). 2017 Jul;124(7):775-798. doi: 10.1007/s00702-016-1661-z. Epub 2017 Feb 24. J Neural Transm (Vienna). 2017. PMID: 28236206 Review.
-
CK2 Regulation: Perspectives in 2021.Biomedicines. 2021 Sep 30;9(10):1361. doi: 10.3390/biomedicines9101361. Biomedicines. 2021. PMID: 34680478 Free PMC article. Review.
Cited by
-
REDOX Balance in Oligodendrocytes Is Important for Zebrafish Visual System Regeneration.Antioxidants (Basel). 2023 Nov 22;12(12):2026. doi: 10.3390/antiox12122026. Antioxidants (Basel). 2023. PMID: 38136146 Free PMC article.
-
Daam2 phosphorylation by CK2α negatively regulates Wnt activity during white matter development and injury.Proc Natl Acad Sci U S A. 2023 Aug 29;120(35):e2304112120. doi: 10.1073/pnas.2304112120. Epub 2023 Aug 22. Proc Natl Acad Sci U S A. 2023. PMID: 37607236 Free PMC article.
-
GABA Receptor Agonists Protect From Excitotoxic Damage Induced by AMPA in Oligodendrocytes.Front Pharmacol. 2022 Jul 26;13:897056. doi: 10.3389/fphar.2022.897056. eCollection 2022. Front Pharmacol. 2022. PMID: 35959434 Free PMC article.
-
Influence of Auditory Integrative Training on Casein Kinase 2 and Its Impact on Behavioral and Social Interaction in Children with Autism Spectrum Disorder.Curr Issues Mol Biol. 2023 May 15;45(5):4317-4330. doi: 10.3390/cimb45050274. Curr Issues Mol Biol. 2023. PMID: 37232743 Free PMC article.
-
Exopolysaccharides isolated from Rhizopus nigricans induced colon cancer cell apoptosis in vitro and in vivo via activating the AMPK pathway.Biosci Rep. 2020 Jan 31;40(1):BSR20192774. doi: 10.1042/BSR20192774. Biosci Rep. 2020. PMID: 31894839 Free PMC article.
References
-
- Axtell R. C., Xu L., Barnum S. R., Raman C. (2006). CD5-CK2 binding/activation-deficient mice are resistant to experimental autoimmune encephalomyelitis: protection is associated with diminished populations of IL-17-expressing T cells in the central nervous system. J. Immunol. 177, 8542–8549. 10.4049/jimmunol.177.12.8542 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous