

A systematic performance evaluation of clustering methods for single-cell RNA-seq data
- PMID: 30271584
- PMCID: PMC6134335
- DOI: 10.12688/f1000research.15666.3
A systematic performance evaluation of clustering methods for single-cell RNA-seq data
Abstract
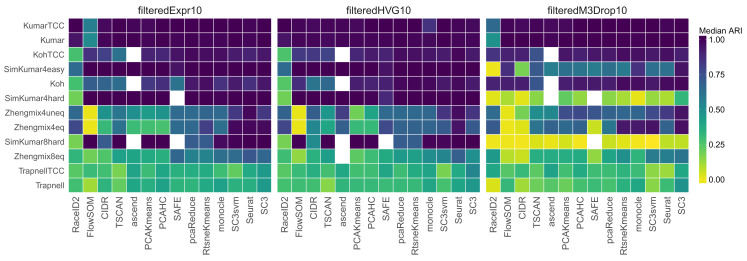
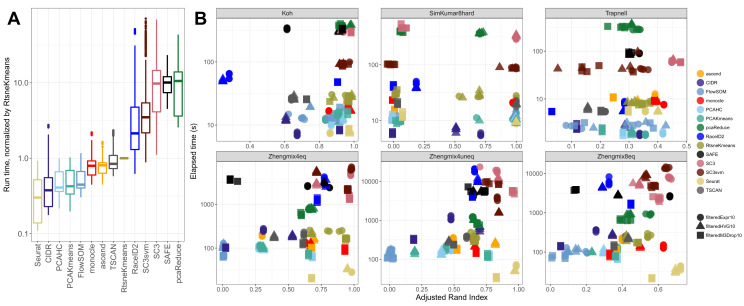
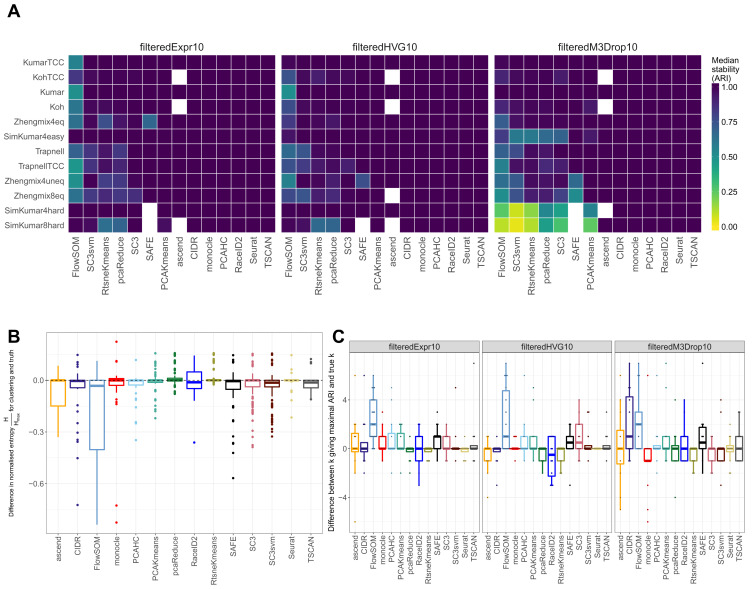
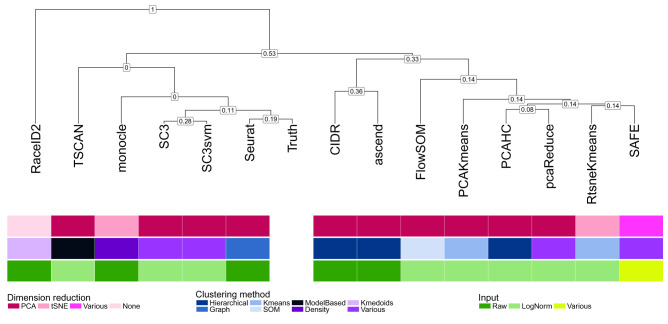
Subpopulation identification, usually via some form of unsupervised clustering, is a fundamental step in the analysis of many single-cell RNA-seq data sets. This has motivated the development and application of a broad range of clustering methods, based on various underlying algorithms. Here, we provide a systematic and extensible performance evaluation of 14 clustering algorithms implemented in R, including both methods developed explicitly for scRNA-seq data and more general-purpose methods. The methods were evaluated using nine publicly available scRNA-seq data sets as well as three simulations with varying degree of cluster separability. The same feature selection approaches were used for all methods, allowing us to focus on the investigation of the performance of the clustering algorithms themselves. We evaluated the ability of recovering known subpopulations, the stability and the run time and scalability of the methods. Additionally, we investigated whether the performance could be improved by generating consensus partitions from multiple individual clustering methods. We found substantial differences in the performance, run time and stability between the methods, with SC3 and Seurat showing the most favorable results. Additionally, we found that consensus clustering typically did not improve the performance compared to the best of the combined methods, but that several of the top-performing methods already perform some type of consensus clustering. All the code used for the evaluation is available on GitHub ( https://github.com/markrobinsonuzh/scRNAseq_clustering_comparison). In addition, an R package providing access to data and clustering results, thereby facilitating inclusion of new methods and data sets, is available from Bioconductor ( https://bioconductor.org/packages/DuoClustering2018).
Keywords: Benchmarking; Clustering; Clustering methods; RNA-seq; Single-cell RNA-seq.
Copyright: © 2020 Duò A et al.
Conflict of interest statement
No competing interests were disclosed.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources