

Mitochondria in Ischemic Stroke: New Insight and Implications
- PMID: 30271667
- PMCID: PMC6147588
- DOI: 10.14336/AD.2017.1126
Mitochondria in Ischemic Stroke: New Insight and Implications
Abstract
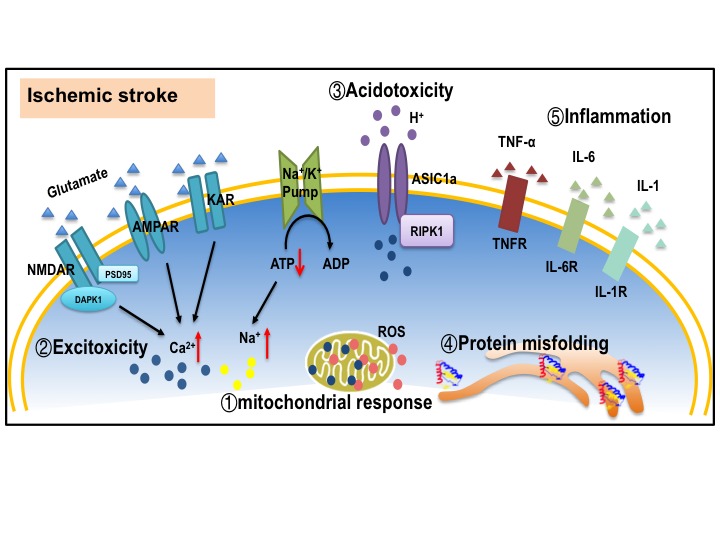
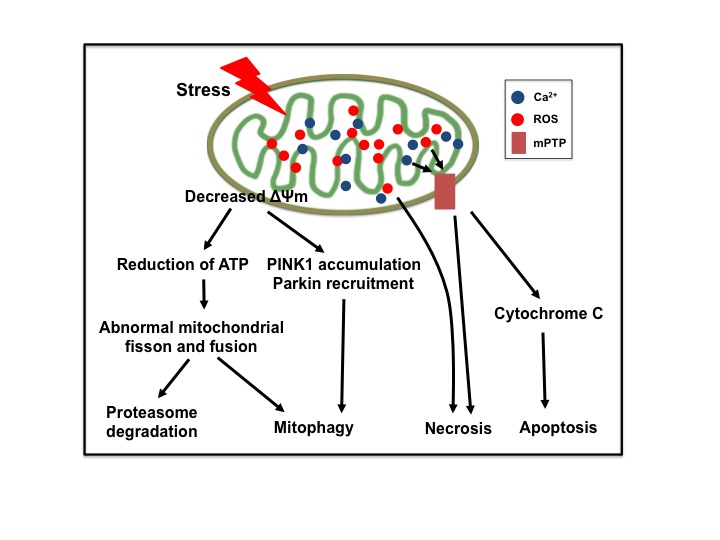
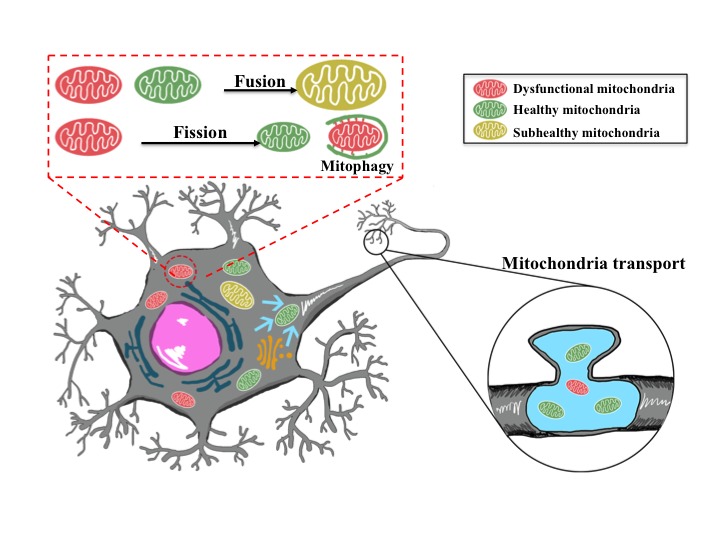
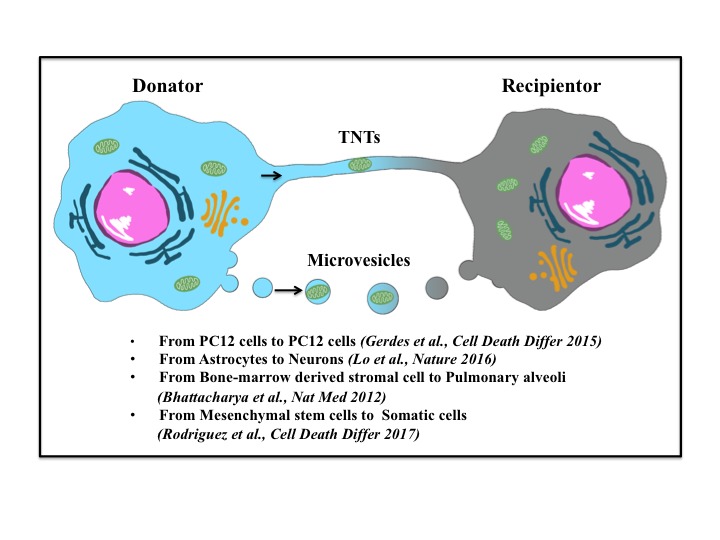
Stroke is the leading cause of death and adult disability worldwide. Mitochondrial dysfunction has been regarded as one of the hallmarks of ischemia/reperfusion (I/R) induced neuronal death. Maintaining the function of mitochondria is crucial in promoting neuron survival and neurological improvement. In this article, we review current progress regarding the roles of mitochondria in the pathological process of cerebral I/R injury. In particular, we emphasize on the most critical mechanisms responsible for mitochondrial quality control, as well as the recent findings on mitochondrial transfer in acute stroke. We highlight the potential of mitochondria as therapeutic targets for stroke treatment and provide valuable insights for clinical strategies.
Keywords: Ischemic stroke; mitochondrial quality control; mitochondrial transfer; mitophagy; neuroprotection.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources