ANIMA: Association network integration for multiscale analysis
- PMID: 30271886
- PMCID: PMC6134339
- DOI: 10.12688/wellcomeopenres.14073.3
ANIMA: Association network integration for multiscale analysis
Abstract
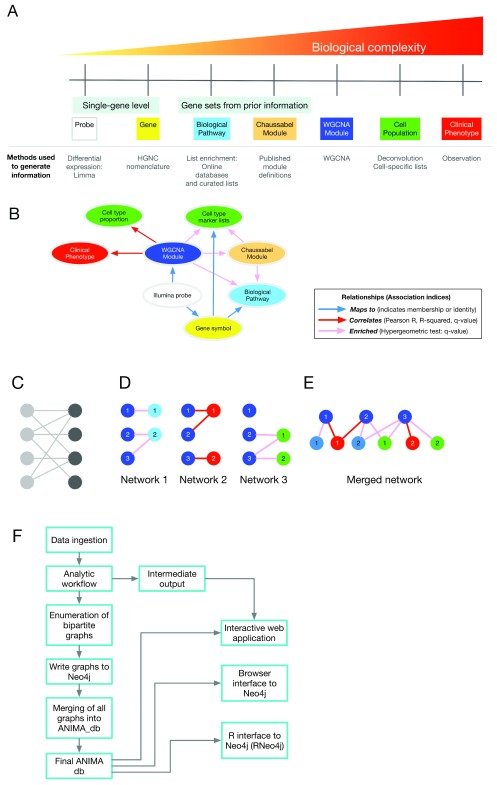
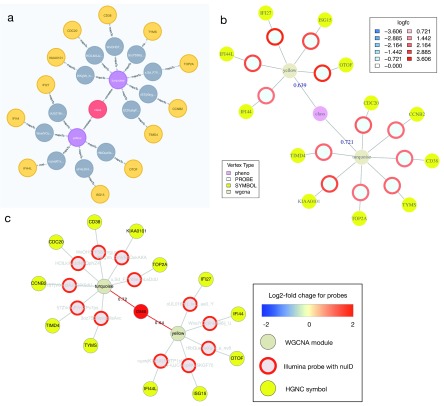
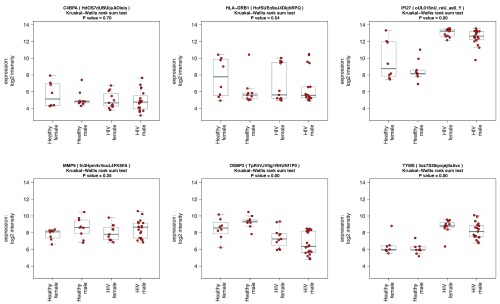
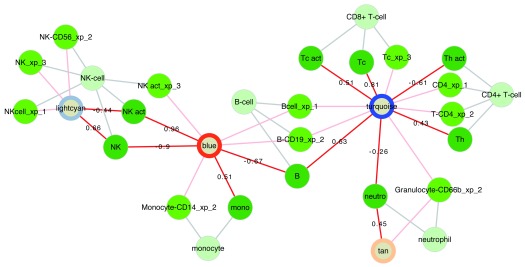
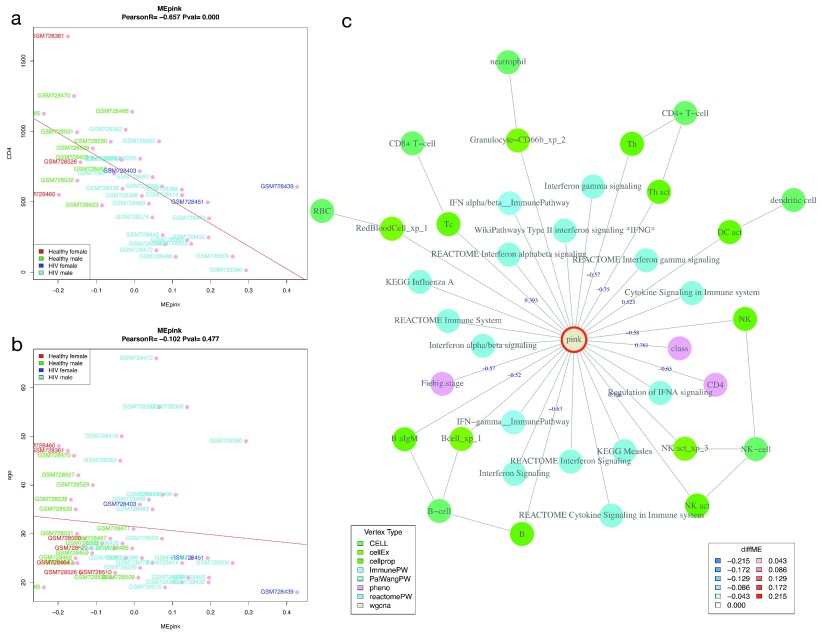
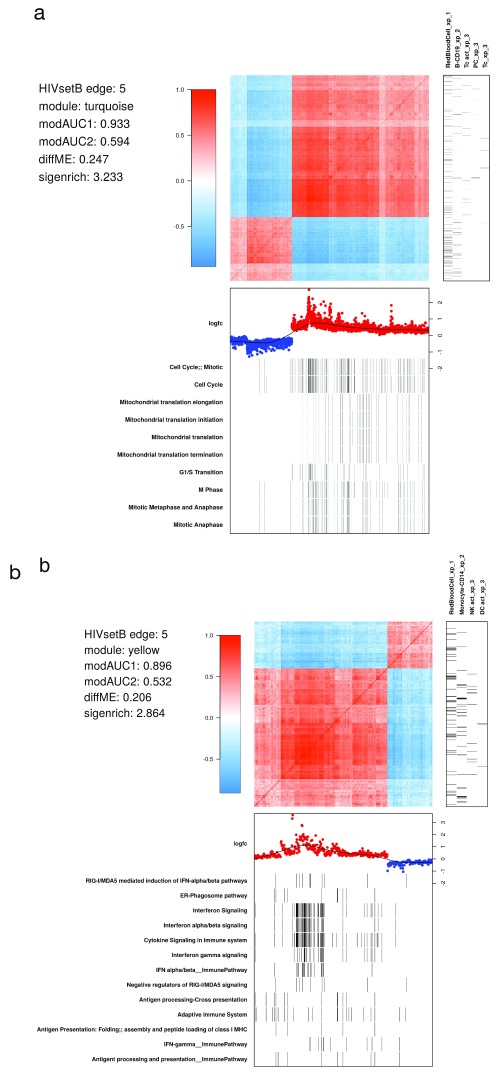
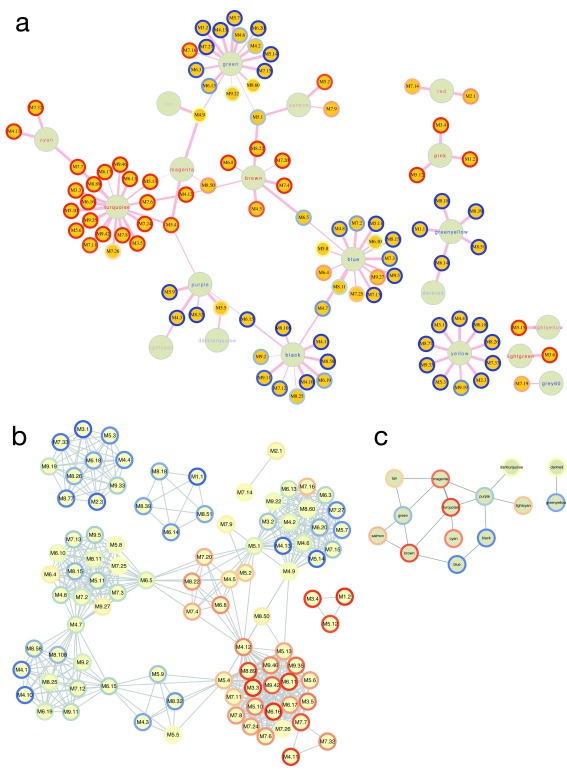
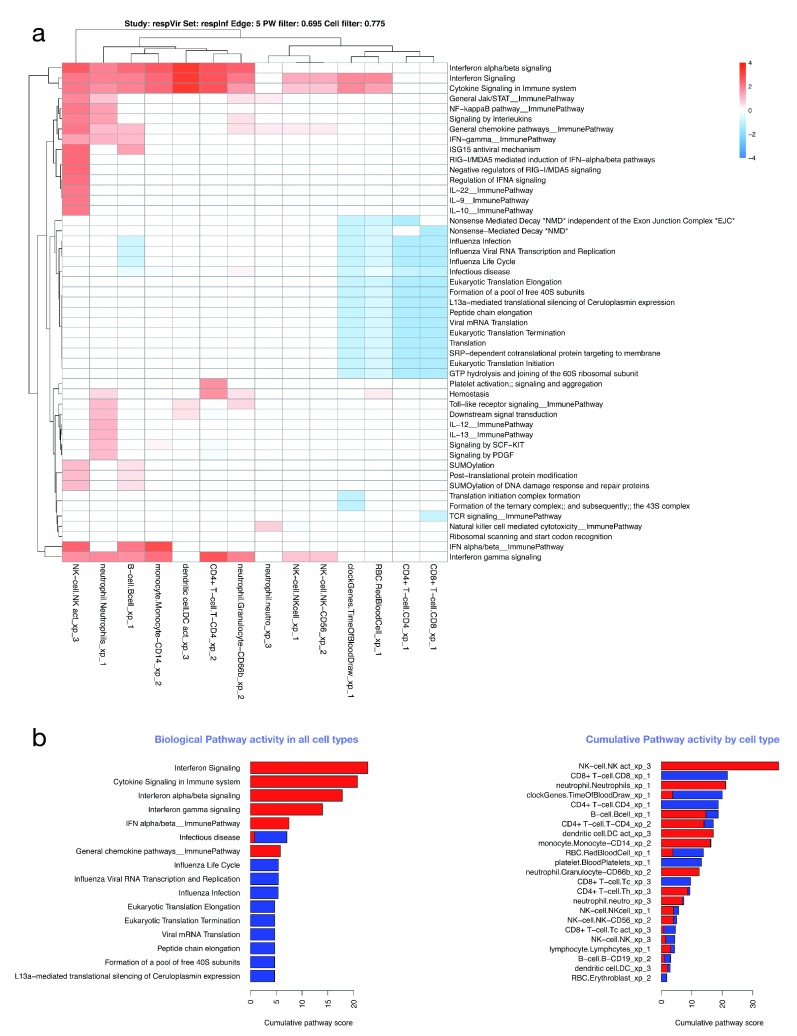
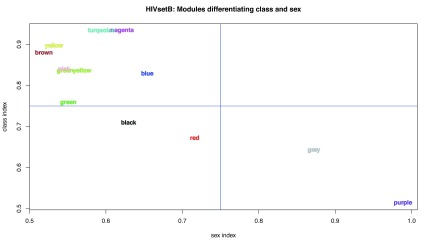
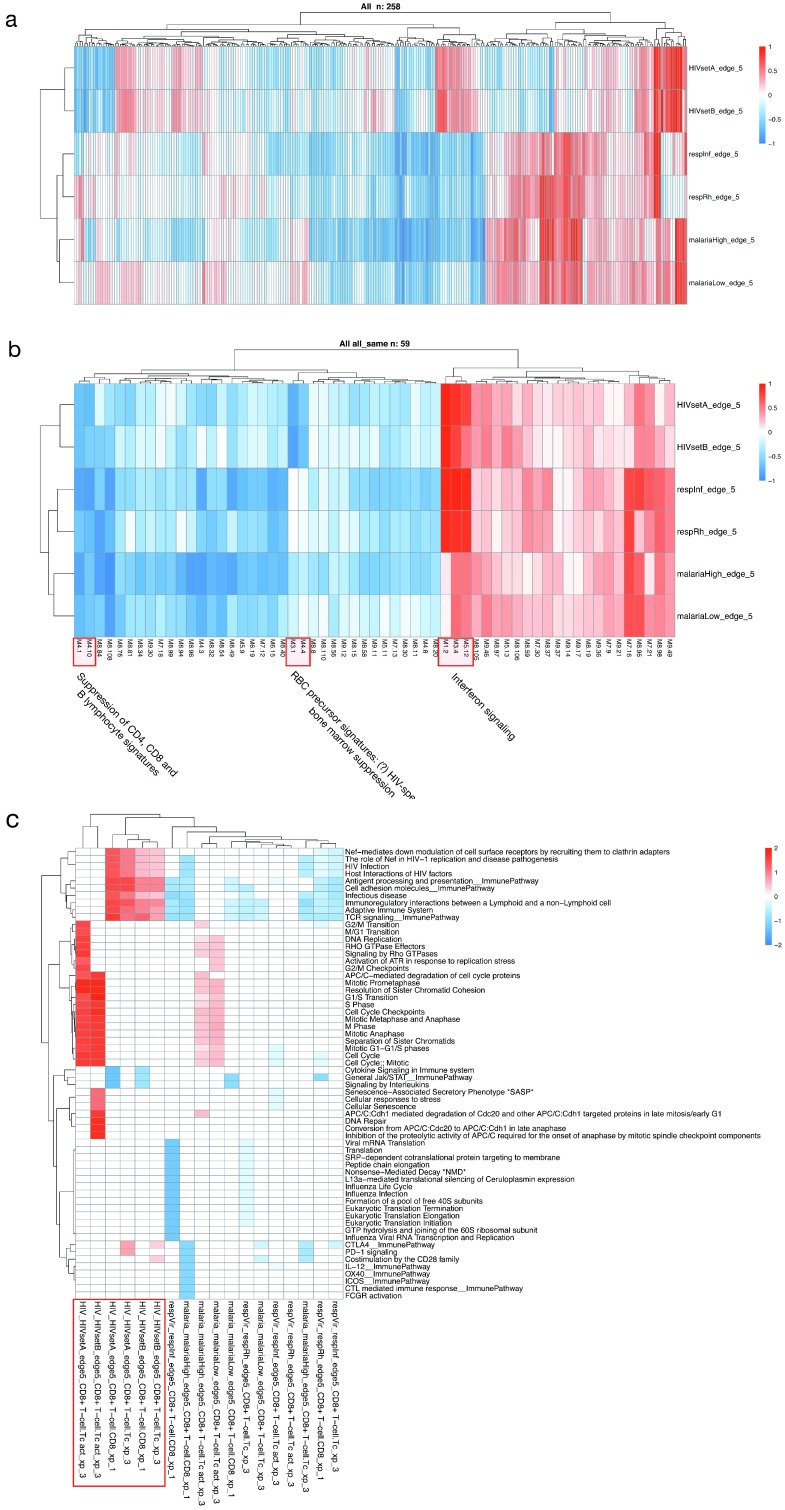
Contextual functional interpretation of -omics data derived from clinical samples is a classical and difficult problem in computational systems biology. The measurement of thousands of data points on single samples has become routine but relating 'big data' datasets to the complexities of human pathobiology is an area of ongoing research. Complicating this is the fact that many publicly available datasets use bulk transcriptomics data from complex tissues like blood. The most prevalent analytic approaches derive molecular 'signatures' of disease states or apply modular analysis frameworks to the data. Here we describe ANIMA (association network integration for multiscale analysis), a network-based data integration method using clinical phenotype and microarray data as inputs. ANIMA is implemented in R and Neo4j and runs in Docker containers. In short, the build algorithm iterates over one or more transcriptomics datasets to generate a large, multipartite association network by executing multiple independent analytic steps (differential expression, deconvolution, modular analysis based on co-expression, pathway analysis) and integrating the results. Once the network is built, it can be queried directly using Cypher (a graph query language), or by custom functions that communicate with the graph database via language-specific APIs. We developed a web application using Shiny, which provides fully interactive, multiscale views of the data. Using our approach, we show that we can reconstruct multiple features of disease states at various scales of organization, from transcript abundance patterns of individual genes through co-expression patterns of groups of genes to patterns of cellular behaviour in whole blood samples, both in single experiments as well in meta-analyses of multiple datasets.
Keywords: Transcriptomics; complex networks; data integration; graph databases.
Conflict of interest statement
No competing interests were disclosed.
Figures