

A multi-amplicon 16S rRNA sequencing and analysis method for improved taxonomic profiling of bacterial communities
- PMID: 30273610
- PMCID: PMC6375304
- DOI: 10.1016/j.mimet.2018.09.019
A multi-amplicon 16S rRNA sequencing and analysis method for improved taxonomic profiling of bacterial communities
Abstract
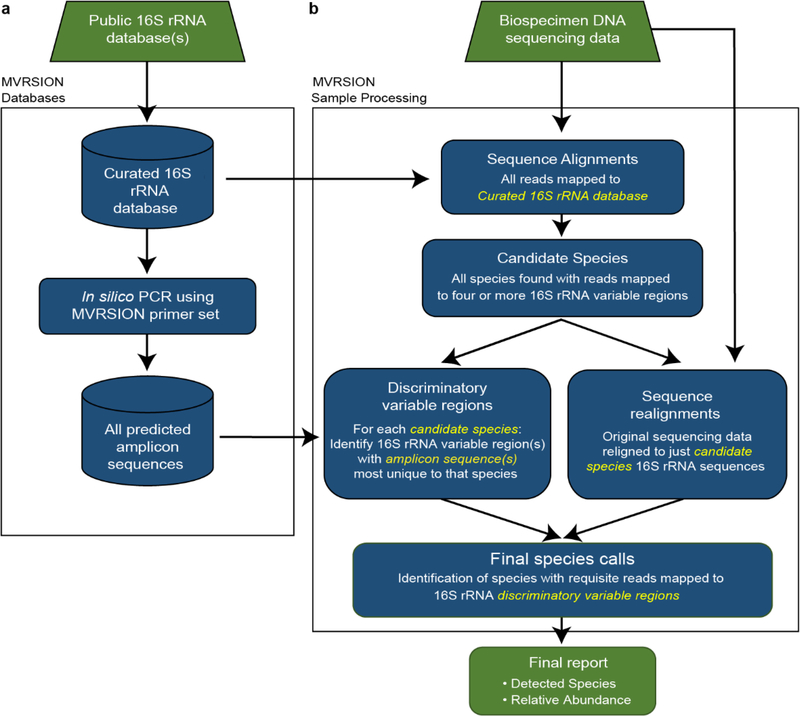
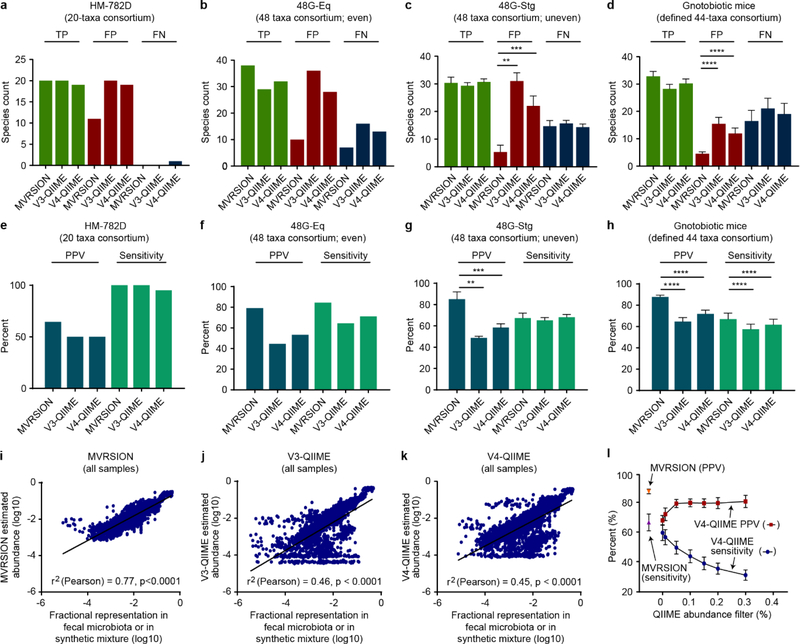
Metagenomic sequencing of bacterial samples has become the gold standard for profiling microbial populations, but 16S rRNA profiling remains widely used due to advantages in sample throughput, cost, and sensitivity even though the approach is hampered by primer bias and lack of specificity. We hypothesized that a hybrid approach, that combined targeted PCR amplification with high-throughput sequencing of multiple regions of the genome, would capture many of the advantages of both approaches. We developed a method that identifies and quantifies members of bacterial communities through simultaneous analysis of multiple variable regions of the bacterial 16S rRNA gene. The method combines high-throughput microfluidics for PCR amplification, short read DNA sequencing, and a custom algorithm named MVRSION (Multiple 16S Variable Region Species-Level IdentificatiON) for optimizing taxonomic assignment. MVRSION performance was compared to single variable region analyses (V3 or V4) of five synthetic mixtures of human gut bacterial strains using existing software (QIIME), and the results of community profiling by shotgun sequencing (COPRO-Seq) of fecal DNA samples collected from gnotobiotic mice colonized with a defined, phylogenetically diverse consortium of human gut bacterial strains. Positive predictive values for MVRSION ranged from 65%-91% versus 44%-61% for single region QIIME analyses (p < .01, p < .001), while the abundance estimate r2 for MVRSION compared to COPRO-Seq was 0.77 vs. 0.46 and 0.45 for V3-QIIME and V4-QIIME, respectively. MVRSION represents a generally applicable tool for taxonomic classification that is superior to single-region 16S rRNA methods, resource efficient, highly scalable for assessing the microbial composition of up to thousands of samples concurrently, with multiple applications ranging from whole community profiling to targeted tracking of organisms of interest in diverse habitats as a function of specified variables/perturbations.
Keywords: 16S rRNA gene; Microbial community analysis; Microbial diversity; Next generation sequencing.
Copyright © 2018 Elsevier B.V. All rights reserved.
Figures
References
-
- Aronesty E (2011). ea-utils : Command-line tools for processing biological sequencing data. Github repository. Retrieved from https://github.com/ExpressionAnalysis/ea-utils
-
- Buffalo V (2011). Scythe - A Bayesian adapter trimmer (Version 0.994). Github repository Retrieved from https://github.com/vsbuffalo/scythe
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
